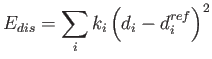UNRES - A PROGRAM FOR COARSE-GRAINED SIMULATIONS OF PROTEINS
Laboratory of Molecular Modeling
Faculty of Chemistry
University of Gdansk
Wita Stwosza 63
80-308 Gdansk, Poland
Scheraga Group
Baker Laboratory of Chemistry
and Chemical Biology
Cornell University
Ithaca, NY 14853-1301, USA
December 4, 2014
1 LICENSE TERMS
-
This software is provided free of charge to academic users, subject to the condition that no part of it be sold or used otherwise for commercial purposes, including, but not limited to its incorporation into commercial software packages, without written consent from the authors. For permission contact Prof. H. A. Scheraga, Cornell University.
-
This software package is provided on an "as is" basis. We in no way warrant either this software or results it may produce.
-
Reports or publications using this software package must contain an acknowledgment to the authors and the NIH Resource in the form commonly used in academic research.
2 CREDITS
The current and former developers of UNRES are listed in this section in alphabetic order together with their current or former affiliations.
Maurizio Chinchio (formerly Cornell Univ., USA)
Cezary Czaplewski (Univ. of Gdansk, Poland)
Carlo Guardiani (Georgia State Univ., USA)
Yi He (Cornell Univ., USA)
Justyna Iwaszkiewicz (Swiss Institute of Bioinformatics, Switzerland)
Dawid Jagiela (Univ. of Gdansk, Poland)
Stanislaw Jaworski (deceased)
Sebastian Kalinowski (Univ. of Gdansk, Poland)
Urszula Kozlowska (deceased)
Pawel Krupa (Univ. of Gdansk, Poland)
Rajmund Kazmierkiewicz (Univ. of Gdansk, Poland)
Jooyoung Lee (Korea Institute for Advanced Studies, Korea)
Adam Liwo (Univ. of Gdansk, Poland)
Mariusz Makowski (Univ. of Gdansk, Poland)
Magdalena Mozolewska (Univ. of Gdansk, Poland)
Marian Nanias (formerly Cornell Univ., USA)
Stanislaw Oldziej (Univ. of Gdansk, Poland)
Jaroslaw Pillardy (Cornell Univ., USA)
Shelly Rackovsky (Mout Sinai School of Medicine, USA)
Daniel Ripoll (formerly Cornell Univ., USA)
Jeff Saunders (Schrodinger Inc., USA)
Harold A. Scheraga (Cornell Univ., USA)
Hujun Shen (Dalian Institute of Chemical Physics, P.R. China)
Adam Sieradzan (Univ. of Gdansk, Poland)
Ryszard Wawak (formerly Cornell Univ., USA)
Tomasz Wirecki (Univ. of Gdansk, Poland)
Marta Wisniewska (Univ. of Gdansk, Poland)
Yanping Yin (Cornell Univ., USA)
Bartlomiej Zaborowski (Univ. of Gdansk, Poland)
Maciej Baranowski (Univ. of Gdansk, Poland)
3 GENERAL INFORMATION
3.1 Purpose
Run coarse-grained calculations of polypeptide chains with the UNRES force field. There are two versions of the package which should be kept separate because of non-overlapping functions: version which runs global optimization (Conformational Space Annealing, CSA) and version that runs coarse-grained molecular dynamics and its extension. Because the installation, input file preparation and running CSA and MD versions are similar, a common manual is provided. Items specific for the CSA and MD version are marked "CSA" and "MD", respectively.
MD version can be used to run multiple-chain proteins (however, that version of the code is a new release and might fail if yet un-checked functions are used). The multi-chain CSA version for this purpose is another package (written largely in C++).
3.2 Functions of the program
-
Perform energy evaluation of a single or multiple conformations (serial and parallel) (CSA and MD).
-
Run canonical mesoscopic molecular dynamics (serial and parallel) (MD).
-
Run replica exchange (REMD) and multiplexing replica exchange (MREMD) dynamics (parallel only) (MD).
-
Run multicanonical molecular dynamics (parallel only) (MD).
-
Run energy minimization (serial and parallel) (CSA and MD).
-
Run conformational space annealing (CSA search) (parallel only) (CSA).
-
Run Monte Carlo plus Minimization (MCM) (parallel only) (CSA).
-
Run conformational family Monte Carlo (CFMC) calculations (CSA).
-
Thread the sequence against a database from the PDB and minimize energy of each structure (CSA).
Energy and force evaluation is parallelized in MD version.
3.3 Companion programs
The structures produced by UNRES can be used as inputs to the following programs provided with this package or separately:
-
xdrf2pdb - converts the compressed coordinate files from MD (but not MREMD)runs into PDB format.
-
-
xdrf2pdb-m - same for MREMD runs (multiple trajectory capacity).
-
-
xdrf2x - converts the plain Cartesian coordinate files into PDB format.
-
-
WHAM - processes the coordinate files from MREMD runs and computes temperature profiles of ensemble averages and computes the probabilities of conformations at selected temperatures; also prepares data for CLUSTER and ZSCORE.
-
-
CLUSTER - does the cluster analysis of the conformations; for MREMD runs takes the coordinate files from WHAM which contain information to compute probabilities of conformations at any temperature.
-
-
PHOENIX - conversion of UNRES conformations to all-atom conformations.
-
-
ZSCORE - force field optimization (for developers).
Please consult the manuals of the corresponding packages for details. Note that not all of these packages are released yet; they will be released depending on their readiness for distribution. Contact Adam Liwo, Cezary Czaplewski or Stanislaw Oldziej for developmental versions of these programs.
3.4 Programming language
This version of UNRES is written almost exclusively in Fortran 77; some subroutines for data management are in ansi-C. The package was parallelized with MPI.
3.5 References
Citing the following references in your work that makes use of UNRES is gratefully acknowledged:
-
[1]
-
A. Liwo, S. Oldziej, M.R. Pincus, R.J. Wawak, S. Rackovsky, H.A. Scheraga. A united-residue force field for off-lattice protein-structure simulations. I: Functional forms and parameters of long-range side-chain interaction potentials from protein crystal data. J. Comput. Chem., 1997, 18, 849-873.
-
[2]
-
A. Liwo, M.R. Pincus, R.J. Wawak, S. Rackovsky, S. Oldziej, H.A. Scheraga. A united-residue force field for off-lattice protein-structure simulations. II: Parameterization of local interactions and determination of the weights of energy terms by Z-score optimization. J. Comput. Chem., 1997, 18, 874-887.
-
[3]
-
A. Liwo, S. Oldziej, R. Kazmierkiewicz, M. Groth, C. Czaplewski. Design of a knowledge-based force field for off-lattice simulations of protein structure. Acta Biochim. Pol., 1997, 44, 527-548.
-
[4]
-
A. Liwo, R. Kazmierkiewicz, C. Czaplewski, M. Groth, S. Oldziej, R.J. Wawak, S. Rackovsky, M.R. Pincus, H.A. Scheraga. United-residue force field for off-lattice protein-structure simulations. III. Origin of backbone hydrogen-bonding cooperativity in united-residue potentials. J. Comput. Chem., 1998, 19, 259-276.
-
[5]
-
A. Liwo, C. Czaplewski, J. Pillardy, H.A. Scheraga. Cumulant-based expressions for the multibody terms for the correlation between local and electrostatic interactions in the united-residue force field. J. Chem. Phys., 2001, 115, 2323-2347.
-
[6]
-
J. Lee, D.R. Ripoll, C. Czaplewski, J. Pillardy, W.J. Wedemeyer, H.A. Scheraga, Optimization of parameters in macromolecular potential energy functions by conformational space annealing. J. Phys. Chem. B, 2001, 105, 7291-7298
-
[7]
-
J. Pillardy, C. Czaplewski, A. Liwo, W.J. Wedemeyer, J. Lee, D.R. Ripoll, P. Arlukowicz, S. Oldziej, Y.A. Arnautova, H.A. Scheraga, Development of physics-based energy functions that predict medium-resolution structures for proteins of the
α, β, and
α/β structural classes. J. Phys. Chem. B, 2001, 105, 7299-7311
-
[8]
-
A. Liwo, P. Arlukowicz, C. Czaplewski, S. Oldziej, J. Pillardy, H.A. Scheraga. A method for optimizing potential-energy functions by a hierarchical design of the potential-energy landscape: Application to the UNRES force field. Proc. Natl. Acad. Sci. U.S.A., 2002, 99, 1937-1942.
-
[9]
-
J. A. Saunders and H.A. Scheraga. Ab initio structure prediction of two α-helical oligomers with a multiple-chain united-residue force field and global search. Biopolymers, 2003, 68, 300-317.
-
[10]
-
J.A. Saunders and H.A. Scheraga. Challenges in structure prediction of oligomeric proteins at the united-residue level: searching the multiple-chain energy landscape with CSA and CFMC procedures. Biopolymers, 2003, 68, 318-332.
-
[11]
-
S. Oldziej, U. Kozlowska, A. Liwo, H.A. Scheraga. Determination of the potentials of mean force for rotation about Cα-Cα virtual bonds in polypeptides from the ab initio energy surfaces of terminally blocked glycine, alanine, and proline. J. Phys. Chem. A, 2003, 107, 8035-8046.
-
[12]
-
A. Liwo, S. Oldziej, C. Czaplewski, U. Kozlowska, H.A. Scheraga. Parameterization of backbone-electrostatic and multibody contributions to the UNRES force field for protein-structure prediction from ab initio energy surfaces of model systems. J. Phys. A, 2004, 108, 9421-9438.
-
[13]
-
S. Oldziej, A. Liwo, C. Czaplewski, J. Pillardy, H.A. Scheraga. Optimization of the UNRES force field by hierarchical design of the potential-energy landscape. 2. Off-lattice tests of the method with single proteins. J. Phys. Chem. B., 2004, 108, 16934-16949.
-
[14]
-
S. Oldziej, J. Lagiewka, A. Liwo, C. Czaplewski, M. Chinchio, M. Nanias, H.A. Scheraga. Optimization of the UNRES force field by hierarchical design of the potential-energy landscape. 3. Use of many proteins in optimization. J. Phys. Chem. B., 2004, 108, 16950-16959.
-
[15]
-
M. Khalili, A. Liwo, F. Rakowski, P. Grochowski, H.A. Scheraga. Molecular dynamics with the united-residue model of polypeptide chains. I. Lagrange equations of motion and tests of numerical stability in the microcanonical mode, J. Phys. Chem. B, 2005, 109, 13785-13797.
-
[16]
-
M. Khalili, A. Liwo, A. Jagielska, H.A. Scheraga. Molecular dynamics with the united-residue model of polypeptide chains. II. Langevin and Berendsen-bath dynamics and tests on model α-helical systems. J. Phys. Chem. B, 2005, 109, 13798-13810.
-
[17]
-
A. Liwo, M. Khalili, H.A. Scheraga. Ab initio simulations of protein-folding pathways by molecular dynamics with the united-residue model of polypeptide chains. Proc. Natl. Acad. Sci. U.S.A., 2005, 102, 2362-2367.
-
[18]
-
F. Rakowski, P. Grochowski, B. Lesyng, A. Liwo, H. A. Scheraga. Implementation of a symplectic multiple-time-step molecular dynamics algorithm, based on the united-residue mesoscopic potential energy function. J. Chem. Phys., 2006, 125, 204107.
-
[19]
-
M. Nanias, C. Czaplewski, H.A. Scheraga. Replica exchange and multicanonical algorithms with the coarse-grained united-residue (UNRES) force field. J. Chem. Theory and Comput., 2006, 2, 513-528.
-
[20]
-
A. Liwo, M. Khalili, C. Czaplewski, S. Kalinowski, S. Oldziej, K. Wachucik, H.A. Scheraga. Modification and optimization of the united-residue (UNRES) potential energy function for canonical simulations. I. Temperature dependence of the effective energy function and tests of the optimization method with single training proteins. J. Phys. Chem. B, 2007, 111, 260-285.
-
[21]
-
U. Kozlowska, A. Liwo, H.A. Scheraga. Determination of virtual-bond-angle potentials of mean force for coarse-grained simulations of protein structure and folding from ab initio energy surfaces of terminally-blocked glycine, alanine, and proline. J. Phys.: Condens. Matter, 2007, 19, 285203.
-
[22]
-
M. Chinchio, C. Czaplewski, A. Liwo, S. Oldziej, H.A. Scheraga. Dynamic formation and breaking of disulfide bonds in molecular dynamics simulations with the UNRES force field. J. Chem. Theory Comput., 2007, 3, 1236-1248.
-
[23]
-
A.V. Rojas, A. Liwo, H.A. Scheraga. Molecular dynamics with the united-residue force field: Ab Initio folding simulations of multichain proteins. J. Phys. Chem. B, 2007, 111, 293-309.
-
[24]
-
A. Liwo, C. Czaplewski, S. Oldziej, A.V. Rojas, R. Kazmierkiewicz, M. Makowski, R.K. Murarka, H.A. Scheraga. Simulation of protein structure and dynamics with the coarse-grained UNRES force field. In: Coarse-Graining of Condensed Phase and Biomolecular Systems., ed. G. Voth, Taylor & Francis, 2008, Chapter 8, pp. 107-122.
-
[25]
-
C. Czaplewski, S. Kalinowski, A. Liwo, H.A. Scheraga. Application of multiplexed replica exchange molecular dynamics to the UNRES force field: tests with α and
α+β proteins. J. Chem. Theory Comput., 2009, 5, 627-640.
-
[26]
-
Y. He, Y. Xiao, A. Liwo, H.A. Scheraga. Exploring the parameter space of the coarse-grained UNRES force field by random search: selecting a transferable medium-resolution force field. J. Comput. Chem., 2009, 30, 2127-2135.
-
[27]
-
U. Kozlowska, A. Liwo. H.A. Scheraga. Determination of side-chain-rotamer and side-chain and backbone virtual-bond-stretching potentials of mean force from AM1 energy surfaces of terminally-blocked amino-acid residues, for coarse-grained simulations of protein structure and folding. 1. The Method. J. Comput. Chem., 2010, 31, 1143-1153.
-
[28]
-
U. Kozlowska, G.G. Maisuradze, A. Liwo, H.A. Scheraga. Determination of side-chain-rotamer and side-chain and backbone virtual-bond-stretching potentials of mean force from AM1 energy surfaces of terminally-blocked amino-acid residues, for coarse-grained simulations of protein structure and folding. 2. Results, comparison with statistical potentials, and implementation in the UNRES force field. J. Comput. Chem., 2010, 31, 1154-1167.
-
[29]
-
A. Liwo, S. Oldziej, C. Czaplewski, D.S. Kleinerman, P. Blood, H.A. Scheraga. Implementation of molecular dynamics and its extensions with the coarse-grained UNRES force field on massively parallel systems; towards millisecond-scale simulations of protein structure, dynamics, and thermodynamics. J. Chem. Theory Comput., 2010, 6, 890-909.
- [30]
- A.K. Sieradzan, U.H.E. Hansmann, H.A. Scheraga, A. Liwo.
Extension of UNRES force field to treat polypeptide chains with D-amino-acid residues.
J. Chem. Theory Comput., 2012, 8, 4746-4757.
- [31]
- P. Krupa, A.K. Sieradzan, S. Rackovsky, M. Baranowski, S. Oldziej,
H.A. Scheraga, A. Liwo, C. Czaplewski.
Improvement of the treatment of loop structures in the UNRES
force field by inclusion of coupling between backbone- and
side-chain-local conformational states
J. Chem. Theory Comput., 2013, 4620-4632.
- [32]
- A.K. Sieradzan, A. Niadzvedtski, H.A. Scheraga, A. Liwo.
Revised backbone-virtual-bond-angle potentials to treat the L- and D-amino
acid residues in the coarse-grained united residue (UNRES) force field.
J. Chem. Theory Comput., 2014, 10, 2194-2203.
4 INSTALLATION
Please follow the instructions in the installation guide to download and put the package on your
system. In what follows, $UNRESROOT is the location of the UNRES package in your system.
It is recommended to install all components of the package using the Cmake utility.
Please follow the instructions in the Installation Guide.
This section describes the installation of only the UNRES component of the package,
using make program. Sample Makefiles are present in the respective source directories.
To produce the executable do the following:
-
(a)
-
To build parallel version, make sure that MPI is installed in your system. Note that the package will have limited functions when compiled in a single-CPU mode. On linux cluster the command source $HOME/.env should be added to .tcshrc or equivalent file to use parallel version of the program, the alternative is to use queuing system like PBS. In some cases the FORTRAN library subroutine GETENV does not work properly with MPI, if the script is run interactively. In such a case try to add the source mygentenv.F and turn on the -DMYGETENV preprocessor flag.
-
(b)
-
Change directory to the respective source directory.
-
(c)
- Select the appropriate Makefile_xxxx or copy the most matching Makefile_xxx
to another name (e.g., Makefile_MySystem) and edit it to customize to your
system. Note that the CSA version works only with MPI.
Makefile_pgf90 - Linux, the pgf90 compiler,
Makefile_intel - Linux, Intel Fortran compiler,
Makefile_gfortran - Gnu Fortran compiler,
Makefile_bluegene - BlueGene/Q (AIX Fortan).
Please note that Makefile must be a symbolic link to the Makefile_xxx of choice. Make sure
that the file cinfo.f is present; if not, execute:
touch cinfo.f
Other systems should not cause problems; all you have to do is to change
the compiler, compiler options, and preprocessor options.
By default, the executables will be placed in $UNRESROOT/bin/unres/CSA
$UNRESROOT/bin/unres/MD and $UNRESROOT/bin/unres/MINIM, respectively.
The following architectures are defined in the .F source files:
-
AIX - AIX systems (put -DAIX as one of the preprocessor options, if this is your system).
-
-
LINUX - Linux (put -DLINUX).
-
-
G77 - Gnu-Fortran compilers (might require sum moderate source code editing) (put -DG77). The recommended compiler is gfortran and not g77.
-
-
PGI - PGI compilers.
-
-
WINPGI - additional setting for PGI compilers for MS Windows.
-
-
SGI - all SGI platforms; should also be good for SUN platforms (put -DSGI).
-
- CRAY - handles some Cray-specific I/Os and other instructions.
-
-
WIN - MS Windows with Digital Fortran compiler (put -DWIN)
For other platforms, the only problems might appear in connection with machine-specific I/O instructions. Many files are opened in the append mode, whose specification in the OPEN statement is quite machine-dependent. In this case you might need to modify the source code accordingly. The other platform dependent routines are the timing routines contained in timing.F. In addition to the platforms specified above, ES9000, SUN, KSR, and CRAY are defined there.
For parallel build -DMP and -DMPI must be set (these are set in Makefile).
IMPORTANT! Apart from this, two define flags: -DCRYST_TOR and -DMOMENT define earlier versions of the force field. The MUST NOT be entered, if the CASP5 and later versions of the force field are used.
-
(d)
-
Build the unres executables by typing at your UNIX prompt:
make # will build unres
make clean # will remove the object files
The bin directory contains pre-built binaries for Red Hat Linux. These executables are specified in the csh scripts listed in section 4.
-
(e)
-
Customize the C-shell scripts unres.unres (to run the parallel version on set of workstation). See the next section of this manual for guidance.
After the executables are build and C-shell scripts customized, you can run the test examples contained in UNRES/examples.
5 CUSTOMIZING YOUR C-SHELL SCRIPT
IMPORTANT NOTE - The unres.csh script is for Linux and should also be easily adaptable to other systems running MPICH. This script is for interactive parallel jobs. Examples of scripts compatible with PBS (pbs.sub) and LoadLever (sp2.sub) queuing systems are also provided.
Edit the following lines in your unres.csh script:
set DD = your_database_directory
e.g., if you installed the package on the directory /usr/local, this line looks like this:
set DD = /usr/local/UNRES/PARAM
set BIN = your_binaries_directory
set FGPROCS = number_of_processors_per_energy/force_evaluation (MD)
e.g., if the root directory is as above:
set BIN = /usr/local/UNRES/bin
6 COMMAND LINE AND FILES
To run UNRES interactively enter the following command at your Unix prompt or put it in the batch script:
unres.csh POTENTIAL INPUT N_PROCS
where:
POTENTIAL specifies the side-chain interaction potential type and must be one of the following:
-
-
LJ - 6-12 radial Lennard-Jones.
-
-
LJK - 6-12 radial Lennard-Jones-Kihara (shifted Lennard Jones).
-
-
BP - 6-12 anisotropic Berne-Pechukas based on Gaussian overlap (dilated Lennard-Jones).
-
-
GB - 6-12 anisotropic Gay-Berne (shifted Lennard-Jones).
-
-
GBV - 6-12 anisotropic Gay-Berne-Vorobjev (shifted Lennard-Jones).
See section 7 (Force Fields) for explanation and usage.
At present, only the LJ and GB potentials are applied. The LJ potential is used in the "CASP3" version of the UNRES force field that is able to predict only α-helical structures. All further version of the UNRES force field use the GB potential. For the description of all above-mentioned potentials see ref. [2].
-
-
INPUT is the prefix for input and output files (see below)
-
-
N_PROCS is the number of processors; for a CSA or REMD/MREMD run it MUST be at least 2.
Note! The script takes one more variable, FGPROCS, as the fourth argument, which is the number of fine-grain processors to parallelize energy evaluations. The corresponding code is in UNRES/CSA, but it was written using MPL instead of MPI and therefore is never used in the present version. At present we have no plans to rewrite fine-grain parallelization using MPI, because we found that the scalability for up to 200 residue polypeptide chains was very poor, due to a small number of interactions and, correspondingly, unfavorable ratio of the overhead to the computation time.
-
-
INPUT.inp contains the main input data and the control parameters of the CSA method.
-
-
INPUT.out_POTENTIAL_xxx is the main output files from different processors; xxx denotes the number of the processor
-
-
INPUT_POTENTIALxxx.stat is the summary files with the energies, energy components, and RMS deviations of the conformations produced by each of the processors; not used in CSA runs; also it outputs different quantity in MD/MREMD runs.
CSA version specific files:
-
-
INPUT_POTENTIALxxx.int is the internal coordinates; in the CSA run
-
-
INPUT_POTENTIAL_000.int contains the coordinates of the conformations, and the other files are empty
-
-
INPUT.CSA.history is the history file from a CSA run. This is an I/O file, because it can be used to restart an interrupted CSA run.
-
-
INPUT.CSA.seed stores the random seed generated in a CSA run; written for restart purposes.
-
-
INPUT.CSA.bank is the current bank of conformations obtained in CSA calculations (expressed as internal coordinates). This information is also stored in INPUT_POTENTIAL000.int
-
-
INPUT.CSA.rbank - as above, but contains random-generated conformations.
MD version specific files:
-
INPUT_MDyyy.pdb is the Cartesian coordinates of the conformations in PDB format.
-
-
INPUT_MDyyy.x is the Cartesian coordinates of the conformations in ASCII format.
-
-
INPUT_MDyyy.cx is the Cartesian coordinates of the conformations in compressed format (need xdr2pdb to convert to PDB format).
The program currently produces some more files, but they are not used for any purposes and most of them are scratched after a run is completed.
The run script also contains definitions of the parameter files through the following environmental variables:
-
SIDEPAR - parameters of the SC-SC interaction potentials (USC SC);
-
-
SCPPAR - parameters of the SC-p interaction potential (USCp); this file can be ignored by specifying the -DOLDSCP preprocessor flag, which means that the built-in parameters are used; at present they are the same as the parameters in the file specified by SCPPAR;
-
-
ELEPAR - parameters of the p-p interaction potentials (Upp);
-
-
FOURIER - parameters of the multibody potentials of the coupling between the backbone-local and backbone-electrostatic interactions (Ucorr);
-
-
THETPAR - parameters of the virtual-bond-angle bending potentials (Ub);
-
-
ROTPAR - parameters of the side-chain rotamer potentials (Urot);
-
-
TORPAR - parameters of the torsional potentials (Urot);
-
-
TORDPAR - parameters of the double-torsional potentials.
-
-
SCCORPAR - parameters of the torsional potentials that account for the coupling between the local backbone and local sidechain states (implemented recently).
7 FORCE FIELDS
UNRES is being developed since 1997 and several versions of the force field were produced. The settings and references to these force fields are summarized below.
Force fields for CSA version (can be used in MD but haven't been parameterized for this purpose).
|
Force field
|
Additional
compiler flags
|
SC-SC
potential
|
Example script
and executables
(Linux; PGF90
and IFC)
|
Structural
classes covered
|
References
|
|
CASP3
|
-DCRYST_TOR
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
-DMOMENT
|
LJ
|
unres_CASP3.csh
unres_pgf90_cryst_tor.exe
unres_ifc6_cryst_tor.exe
|
only α
|
[1,2,4]
|
|
ALPHA
|
-DMOMENT
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
|
GB
|
unres_CASP4.csh
unres_pgf90_moment.exe
unres_ifc6_moment.exe
|
only α
|
[5,6,7]
|
|
BETA
|
-DMOMENT
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
|
GB
|
unres_CASP4.csh
unres_pgf90_moment.exe
unres_ifc6_moment.exe
|
only β
|
[5,6,7]
|
|
ALPHABETA
|
-DMOMENT
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
|
GB
|
unres_CASP4.csh
unres_pgf90_moment.exe
unres_ifc6_moment.exe
|
all
|
[5,6,7]
|
|
CASP5
|
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
|
GB
|
unres_CASP5.csh
unres_pgf90.exe
unres_ifc6.exe
|
all
|
[8,9,10,12]
|
|
3P
|
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
|
GB
|
unres_3P.csh
unres_pgf90.exe
unres_ifc6.exe
|
all
|
[13,14]
|
|
4P
|
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
|
GB
|
unees_4P.csh
unres_pgf90.exe
unres_ifc6.exe
|
all
|
[13,14]
|
Force fields for MD version [16,17].
|
Force field
|
Additional
compiler flags
|
SC-SC
potential
|
Example script
and executables
(Linux; PGF90
and IFC)
|
Structural
classes covered
|
References
|
|
GAB
|
-DCRYST_BOND
-DCRYST_THETA
-DCRYST_SC
-DPROCOR
|
GB
|
unres_GAB.csh
|
mostly α
|
[20]
|
|
E0G
|
-DCRYST_BOND
-DCRYST_THET
-DCRYST_SC
-DPROCOR
|
GB
|
unres_E0G.csh
|
mostly α
|
[20]
|
|
E0LL2Y
|
-DPROCOR
|
GB
|
unres_ab.csh
|
all
|
[20,21,26,27,28]
|
The example scripts (the *.csh filed) contain all appropriate parameter files, while the energy-term weights are provided in the example input files listed in EXAMPLES.TXT (*.inp; see section 8. for description of the input files). However, it is user's responsibility to specify appropriate compiler flags. Note that a version WILL NOT work, if the force-field specific compiler flags are not set. The parameter files specified in the run script also must strictly correspond to the energy-term weights specified in the input file. The parameter files for specific force fields are also specified below and the energy-term weights are specified in section 8.
The parameter files are as follows (the environment variables from section 6 are used to identify the parameters):
CASP3:
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_cryst.parm |
|
TORDPAR |
torsion_double_631Gdp.parm (not used) |
|
SIDEPAR |
scinter_LJ.parm |
|
ELEPAR |
electr.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_GAP.parm (not used) |
|
SCCORPAR |
scor_am1_pawel.dat (not used) |
ALPHA, BETA, ALPHABETA (CASP4):
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_ecepp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm (not used) |
|
SIDEPAR |
scinter_GB.parm |
|
ELEPAR |
electr.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_GAP.parm |
|
SCCORPAR |
sccor_am1_pawel.dat (not used) |
CASP5:
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_631Gdp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm |
|
SIDEPAR |
scinter_GB.parm |
|
ELEPAR |
electr_631Gdp.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_opt.parm.1igd_iter7n_c |
|
SCCORPAR |
sccor_am1_pawel.dat (not used) |
3P:
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_631Gdp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm |
|
SIDEPAR |
sc_GB_opt.3P7_iter81_1r |
|
ELEPAR |
electr_631Gdp.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_opt.parm.1igd_hc_iter3_3 |
|
SCCORPAR |
sccor_am1_pawel.dat (not used) |
4P:
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_631Gdp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm |
|
SIDEPAR |
sc_GB_opt.4P5_iter33_3r |
|
ELEPAR |
electr_631Gdp.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_opt.parm.1igd_hc_iter3_3 |
|
SCCORPAR |
sccor_am1_pawel.dat (not used) |
GAB:
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_631Gdp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm |
|
SIDEPAR |
sc_GB_opt.1gab_3S_qclass5no310-shan2-sc-16-10-8k |
|
ELEPAR |
electr_631Gdp.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_opt.parm.1igd_hc_iter3_3 |
|
SCCORPAR |
sccor_pdb_shelly.dat |
E0G:
|
BONDPAR |
bond.parm |
|
THETPAR |
thetaml.5parm |
|
ROTPAR |
scgauss.parm |
|
TORPAR |
torsion_631Gdp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm |
|
SIDEPAR |
sc_GB_opt.1e0g-52-17k-2k-newclass-shan1e9_gap8g-sc |
|
ELEPAR |
electr_631Gdp.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_opt.parm.1igd_hc_iter3_3 |
|
SCCORPAR |
sccor_pdb_shelly.dat |
E0LL2Y:
|
BONDPAR |
bond_AM1.parm |
|
THETPAR |
theta_abinitio.parm |
|
ROTPAR |
rotamers_AM1_aura.10022007.parm |
|
TORPAR |
torsion_631Gdp.parm |
|
TORDPAR |
torsion_double_631Gdp.parm |
|
SIDEPAR |
scinter_$POT.parm |
|
ELEPAR |
electr_631Gdp.parm |
|
SCPPAR |
scp.parm |
|
FOURIER |
fourier_opt.parm.1igd_hc_iter3_3 |
|
SCCORPAR |
sccor_am1_pawel.dat |
Additionally, for E0LL2Y, the following environment variables and files are required to generate random conformations:
THETPARPDB thetaml.5parm
ROTPARPDB scgauss.parm
For CSA, the best force field is 4P. For MD, the E0LL2Y force field is best for ab initio prediction but provides medium resolution (5 A for 60-residue proteins) and overemphasizes β-structures and has to be run with secondary-structure-prediction information. For prediction of the structure of mostly α-protein, and for running dynamics of large proteins, the best is the GAB force field. All these force fields were trained by using our procedure of hierarchical optimization [13,14]. The 4P and E0LL2Y force fields have considerable power independent of structural class. The ALPHA, BETA, and ALPHABETA force fields (for CSA) were used in the CASP4 exercises and the CASP5 force field was used in the CASP5 exercise with some success; ALPHA predicts reasonably the structure of α-helical proteins and is still not obsolete, while for β- and
α + β-structure prediction 3P or 4P should be used, because they are cheaper and more reliable than BETA and ALPHABETA. The early CASP3 force field is included for historical reasons only.
8 INPUT FILES
8.1 Main input data file
Most of the data are organized as data lists, where the data can be put in any order, using a series of statements of the form:
KEYWORD=value
for simple non-logical variables
or just
KEYWORD
to indicate that the corresponding option is turned on. For array variables the assignment statement is:
KEYWORD=value1,value2,...
However, the data lists are unnamed and that must be placed EXACTLY in the order indicated below. The presence of an & in the 80th column of a line indicates that the next line will belong to the same data group. The parser subroutines that interpret the keywords are case insensitive.
Each group of data organized as a data list is indicated as data list format input.
8.1.1 Title
Any string containing up to 80 characters. The first input line is always interpreted as title.
8.1.2 Control data
This data section is in data list format and is read in the READ_CONTROL subroutine.
-
-
TIMLIM - time limit in minutes (960)
-
-
MINIMIZE - if present, energy minimization will be carried out.
-
-
REGULAR - regularize the read in conformation (usually a crystal or NMR structure) by doing a series of three constrained minimizations, to keep the structure as close as possible to the starting (experimental) structure. The constraints are the CA-CA distances of the initial structure. The constraints are gradually diminished and removed in the last minimization.
-
-
SOFTREG - regularize the read in conformation (usually a crystal or NMR structure) by doing a series of constrained minimizations, with additional use of soft potential and secondary structure freezing, to keep the structure as close as possible to the starting (experimental) structure.
-
-
CSA - if present, the run is a CSA run. At present, this is the only reliable mode of doing global conformational search with this package; it is NOT recommended to use MCM or THREAD for this purpose.
-
-
MCM - if present, this is a Monte Carlo Minimization (MCM) run.
-
-
MULTCONF - if present, conformations will be read from the INPUT.intin file.
-
-
MD - run canonical MD (single or multiple trajectories).
-
-
RE - run REMD or MREMD (parallel jobs only).
-
-
MUCA - run multicanonical MD calculations (parallel jobs only).
-
-
MAP=number (integer) - Conformational map will be calculated in chosen angles.
-
-
THREAD=number (integer) - Threading or threading-with-minimization run, using a database of structures contained in the $DD/patterns.cart pattern data base (502 chains or chain fragments), using a total number patterns. It is recommended to use this with energy minimization; this implies regularization of each minimized pattern. See refs. [2] and [3].
-
-
CHECKGRAD - compare numerical and analytical gradient; to be followed by:
-
-
CART - energy gradient in virtual-bond vectors (Cartesian coordinates)
-
-
INT - energy gradient in internal coordinates (default)
-
-
CARINT - derivatives of the internal coordinates in the virtual-bond vectors.
-
-
ONE_LETTER - one-letter and not three-letter code of the amino-acid residues is used.
-
-
SYM (1) - number of chains with same sequence (for oligomeric proteins only).
-
-
PDBSTART - the initial conformation is read in from a PDB file.
-
-
UNRES_PDB - the starting conformation is in UNRES representation (Cα and SC coordinates only). This keyword MUST appear in such a case or the program will generate erroneous and unrealistic side-chain coordinates.
-
-
RAND_CONF - start from a random conformation.
-
-
EXTCONF - start from an extended chain conformation.
-
-
PDBOUT - if present, conformations will be output in PDB format. Note that this keyword affects only the output from single energy evaluation, energy minimization and multiple-conformation data. To request conformations from MD/MREMD runs in PDB format, the MDPDB keyword must be placed on the MD input record.
-
-
MOL2OUT - if present, conformations will be output in SYBYL mol2 format.
-
-
REFSTR - if present, reference structure will be read (e.g., to monitor the RMS deviation from the crystal structure).
-
-
PDBREF - if present, a reference structure will be read in to compare the calculated conformations with it.
-
-
UNRES_PBD - the starting/reference structure is read from an UNRES-generated PDB file.
Keywords: PDBOUT, MOL2OUT, PDBREF, and PDBSTART are ignored for a CSA run. Output mode for MD version is specified in MD input (see section 8.1.6).
-
-
CONSTR_DIST=number
-
-
0 - no distance restraints,
-
> 0 - imposes harmonic restraints on selected distances; see section 5.12. In MD version, also restraints on the q variable [20] can be used.
-
-
WEIDIS=number (real) the weight of the distance term; applies for REGULARIZE and THREAD, otherwise ignored.
-
-
USE_SEC_PRED - use secondary-structure prediction information.
-
-
SEED=number (integer) (no default) Random seed (required, even if the run is not a CSA, MCM, MD or MREMD run).
-
-
PHI - only the virtual-bond dihedral angles γ are considered as variables in energy minimization.
-
-
BACK - only the backbone virtual angles (virtual-bond angles theta and virtual-bond dihedral angles γ) are considered as variables in energy minimization.
By default, all internal coordinates: θ, γ, and the side-chain centroid polar angles α and β are considered as variables in energy minimization.
-
-
RESCALE_MODE=number (real) Choice of the type of temperature dependence of the force field.
-
-
0 - no temperature dependence
-
1 - homographic dependence (not implemented yet with any force field)
-
2 - hyperbolic tangent dependence [20].
-
-
T_BATH=number (real) temperature (for MD runs and temperature-dependent force fields).
The following keywords apply to MCM only:
-
MAXGEN=number (integer) (10000) maximum number of conformations generated in a single MCM iteration
-
-
MAXOVERLAP=number (integer) (1000) maximum number of conformations with "bad" overlaps allowed to appear in a row in a single MCM iteration.
-
-
DISTCHAINMAX - (multi-chain capacity only) maximum distance between the last residue of a given chain and the first residue of the next chain such that restraints will not be imposed; quartic restraints will be imposed for greater distances.
-
-
ENERGY_DEC - detailed energies will be printed for each interacting pair or each virtual bond, virtual-bond angle and dihedral angle, side chain, etc. DO NOT use unless a single energy evaluation was requested.
This data section is in data list format and is read in the READ_MINIM subroutine.
This data group is present, if MINIMIZE was specified on the control card. Otherwise, it must not appear.
-
CART - minimize in virtual-bond vectors instead of angles.
-
-
MAXMIN=number (integer) (2000) maximum number of iterations of the SUMSL minimizer.
-
-
MAXFUN=number (integer) (5000) maximum number of function evaluations in a single minimization.
-
-
TOLF=number (real) (1.0e-2) Tolerance on function.
-
-
RTOLF=number (real) (1.0d-4) Relative tolerance on function.
-
- PRINT_INI - turns on printing nondefault minimization parameters,
initial variables, and gradients in the SUMSL procedures.
-
- PRINT_FINAL - turns on printing final variables and gradients in
SUMSL.
-
- PRINT_STAT - turns on printing abbreviated minimization protocol.
The SUMSL minimizer is used in UNRES/CSA. For detailed description of the control parameters see the source file cored.f and sumsld.f
8.1.4 CSA control parameters
This data group should be present only, if CSA was specified on the control card. It is recommended that the readers to read publications on CSA method for more complete description of the parameters. Brief description of parameters:
-
NCONF=number (integer) (50) This corresponds to the size of the bank at the beginning of the CSA procedure. The size of the bank, nbank, is set to nconf. If necessary (at much later stages of the CSA: see icmax below), nbank increases by multiple of nconf.
-
-
JSTART=number (integer) (1)
-
-
JEND=number (integer) (1) This corresponds to the limit values of do loop, each of which corresponds to an separate CSA run. If jstart=1, and jstart=100, this routine will repeat 100 separate CSA runs (limited by CPU) each one with separate random number initialization. The only difference between two CSA runs (one with jstart=jend=1 and another one with jstart=jend=2) would be different random number initializations if other parameters are identical.
-
-
NSTMAX=number (integer) (500000) This is to set a limit the total number of local minimizations of CSA before termination.
N1=number (integer) (6)
N2=number (integer) (4)
N3=number (integer) (0)
N4=number (integer) (0)
N5=number (integer) (0)
N6=number (integer) (10)
N7=number (integer) (0)
N8=number (integer) (0)
N9=number (integer) (0)
IS1=number (integer) (1)
IS2=number (integer) (8)
These numbers are used to generate trial conformations for each seed. See the file newconf.f for more details.
-
n1: the total number of trial conformations for each seed by substituting nran number of variable angles (see subroutine newconf1ab and subroutine newconf1ar),
-
n2: the total number of trial conformations for each seed by substituting nran number of groups of variable angles (see subroutine newconf1bb and subroutine newconf1br),
-
n3: the total number of trial conformations for each seed by substituting a window of residues which forms a β-hairpin, if there is no enough β-hairpins uses the same algorithm as n6,
-
n4: the total number of trial conformations for each seed by shifting the turn in β-hairpin by +/- 1 or 2 residues, if there is no enough β-hairpins uses the same algorithm as n6,
-
n5: not used,
-
n6: the total number of trial conformations for each seed by substituting a window of residues [is1,is2] inclusive. The size of the window is determined in a random fashion (see subroutine newconf_residue for generation of the trial conformations),
-
n7: the total number of trial conformations for each seed by copying a remote strand pair forming nonlocal β-sheet contact,
-
n8: the total number of trial conformations for each seed by copying an α-helical segment,
-
n9: the total number of trial conformations for each seed by shifting the α-helical segment by +/- 1 or 2 residues.
Typical values used for a 75-residue helical protein is (6 4 0 0 0 10 1 26) for (n1,n2,n3,n4,n5,n6,is1,is2), respectively. In this example, a total of 20 trial conformations are generated for a seed Usually is1=1 is used for all applications, and the value of is2 is set about to 1/3 of the total number of residues. n3, n4 and n7 are design to help in case of proteins with β-sheets
NRAN0=number (integer) (4)
NRAN1=number (integer) (2)
IRR=number (integer) (1)
These numbers are used to determine if the CSA stage is very early. One can use (4 2 1) for these values. For more details one should look into the file, newconf.f, for more details.
NTOTAL=number (integer) (10000)
CUT1=number (real) (2.0)
CUT2=number (real) (5.0)
Annealing schedule is set in following fashion. The value of D_cut is reduced geometrically from 1/cut1 of D_ave (at the beginning) to 1/cut2 of D_ave (after ntotal number of minimizations) where D_ave is the average distance between two conformations in the First_bank.
-
-
ESTOP=number (real) (-3000.0) The CSA procedure stops if a conformations with energy lower than estop is obtained. If the do-loop set by jstart and jend requires more than one loop, the program will go on until the do-loop is finished.
-
-
ICMAX=number (integer) (3) The maximum value of cycle (see the original publications for details). If the number of cycle exceeds this value the program will add nconf more conformations to Bank and First_bank to continue CSA procedure if the new size of the nbank is within the maximum set by nbankm (see above). If the size of nbank exceeds the maximum set by nbankm the CSA procedure for this run will stop and next CSA will begin depending on the do-loop set by jstart and jend.
-
-
IRESTART=number (integer) (0) This tells you if the run is fresh start (irestart=0) or a restart (irestart=1) starting from an old results
-
-
NDIFF=number (integer) (2) The number of variables use in comparison when structure is added to the bank, 4 - all angles, 2 - only backbone angles γ and θ
-
-
NBANKTM=number (integer) (0) The maximum number of structures saved in *.CSA.bankt as history of the run Do not use bankt on massively parallel computation as it kills scalability.
-
-
DELE=number (real) (20.0) Energy cutoff for bankt.
-
-
DIFCUT=number (real) (720.0) Angle cutoff for bankt.
-
-
IREF=number (integer) (0) 0 - normal run, 1 - local CSA which generates only structures close to the reference one read from *.CSA.native.int file.
-
-
RMSCUT=number (real) (4.0) CA RMSD cut off used in local CSA
-
-
PNCCUT=number (real) (0.5) Percentage of native contact used in local CSA
-
-
NCONF_IN=number (integer) (0) The number of conformation read for the first bank from the input file *.intin
Optionally, the CSA parameters can be read from file INPUT.CSA.in, if this file exists. If so, they are read in free format in the following order:
nconf
jstart,jend
nstmax
n1,n2,n3,n4,n5,n6,n7,n8,is1,is2
nran0,nran1,irr
nseed
ntotal,cut1,cut2
estop
icmax,irestart
ntbankm,dele,difcut
iref,rmscut,pnccut
ndiff
8.1.5 MCM data
(Data list format, subroutine MCMREAD.)
This data group is present, if MCM was specified on the control card. Otherwise it must not appear.
-
MAXACC=number (integer) (100) Maximum number of accepted conformations.
-
-
MAXTRIAL=number (integer) (100) Maximum number of unsuccessful trials in a row.
-
-
MAXTRIAL_ITER=number (integer) (1000) Maximum number of unsuccessful trials in a single iteration.
-
-
MAXREPM=number (integer) (200) Maximum number of repetitions of the same minimum.
-
-
RANFRACT=number (real) (0.5d0) Fraction of chain-rebuild motions.
-
-
OVERLAP=number (real) (1.0d3) Bad contact energy criterion.
-
-
NSTEPH=number (integer) (0) Number of heating step in adaptive sampling.
-
-
NSTEPC=number (integer) (0) Number of cooling step in adaptive sampling.
-
-
TMIN=number (real) (298.0d0) Minimum temperature in adaptive-temperature sampling).
-
-
TMAX=number (real) (298.0d0) Maximum temperature in adaptive-temperature sampling).
The temperature is changed according to the formula:
T = TMIN*EXP(ISTEPH*(TMAX-TMIN)/NSTEPH) when heating
and
T = TMAX*EXP(-ISTEPC*(TMAX-TMIN)/NSTEPC) when cooling
The default is to use a constant temperature.
-
-
NWINDOW=number (integer) (0) Number of windows in which the variables will be perturbed; the windows are defined by the numbers of the respective amino-acid residues. If NWINDOW is nonzero, after specifying all MCM input the next lines must define the windows. Each line looks like this:
winstart winend (free format)
e.g. if NWINDOW=2, the input:
4 10
15 20
will mean that only the variables of residues 4-10 and 15-20 will be perturbed. However, in general, all variables will be considered in minimization.
-
-
PRINT_MC=number (0) Printout level in MCM. 0 - no intermediate printing, 1 and 2 - moderate printing, 3 - extensive printing.
-
-
NO_PRINT_STAT - no output to INPUT_POTENTIALxxx.stat.
-
-
NO_PRINT_INT - no internal-coordinate output to INPUT_POTENTIALxxx.int.
8.1.6 MD data
(Mixed format; subroutine READ_MDPAR.)
-
NSTEP (1000000) number of time steps per trajectory.
-
-
NTWE (100) NTWX (1000) frequency of energy and coordinate output, respectively. The coordinates are dumped in the pdb or compressed Gromacs (cx) format, depending on the next keyword. NTWE=0 means no energy dump.
-
-
MDPDB - dump coordinates in the PDB format (cx otherwise)
-
-
TRAJ1FILE only the master processor outputs coordinates. This feature pertains only to REMD/MREMD jobs and overrides NTWX; coordinates are dumped at every exchange in MREMD.
-
-
REST1FILE only the master writes the restart file
-
-
DT (real) (0.1) time step; the unit is "molecular time unit" (mtu); 1 mtu = 48.9 fs
-
-
DAMAX (real) (1.0) maximum allowed change of acceleration during a single time step. The time step gets scaled down, if this is exceeded.
-
-
DVMAX (real) (20.0) - maximum allowed velocity (in A/mtu)
-
-
EDRIFTMAX (real) (10.0) - maximum allowed energy drift in a single MD step (10 kcal/mol)
-
-
REST - restart flag. The calculation is restarted if present.
-
-
LARGE - very detailed output. Don't use except for debugging.
-
-
PRINT_COMPON - prints energy components.
-
-
RESET_MOMENT (1000) - frequency of zeroing out the total angular momentum when running Berendsen mode calculations (for Langevin calculations meaningless).
-
-
RESET_VEL=number (integer) (1000) - frequency of resetting velocities to values from Gaussian distribution.
-
-
RATTLE - use the RATTLE algorithm (constraint bonds); not yet implemented.
-
-
RESPA - use the Multiple Time Step (MTS) or Adaptive Multiple Time Step (A-MTS) algorithm [18]. Without this flag the variable time step (VTS) [16] is run.
-
-
NTIME_SPLIT=number (integer) (1) - initial number of time-split steps
-
-
MAXTIME_SPLIT=number (integer) (64) - maximum number of time-split step
If NTIME_SPLIT==MAXTIME_SPLIT, MTS is run.
-
-
R_CUT=number (real) (2.0) - the cut-off distance in splitting the forces into short- and long-range in site-site VDW distance units.
-
-
LAMBDA (real) (0.3) - the transition length (in site-site VDW distance units) between short- and long-range forces.
-
-
XIRESP - flag to use MTS/A-MTS with Nosé-Hoover/Nosé-Poincaré thermostats.
-
-
LANG=number (integer) (0) Langevin dynamics flag:
-
-
0 - No explicit Langevin dynamics.
-
1 - Langevin with direct integration of the equations of motion (recommended for Langevin calculations)
-
2 - Langevin calculation with analytical pre-integration of the friction and stochastic part of the equations of motion using an algorithm adapted from TINKER. This is MUCH MORE time- and memory-consuming than 1 and requires compiling without the -DLANG0 flag and enormously increases memory requirements.
-
3 - The stochastic integrator developed by Cicotti and coworkers.
-
4 - for other stochastic integrators (not used at present).
Note: With the enclosed code, the -DLANG0 compiler flag is included which disables LANG=2 and LANG=3
-
-
TBF - Berendsen thermostat.
-
-
TAU_BATH (1.0) (units are mtus; 1mtu=48.9 fs) - constant of the coupling to the thermal bath used with the Berendsen thermostat.
-
-
NOSEPOINCARE99 - the Nose-Poincare thermostat as of 1999 will be used.
-
-
NOSEPOINCARE01 - the Nose-Poincare thermostat as of 2001 will be used.
-
-
NOSEHOOVER96 - the Nose-Hoover thermostat will be used.
-
-
Q_NP=number (real) (0.1) - the value of the mass of the fictitious particle in the calculations with the Nose-Poincare thermostat.
-
-
T_BATH (300.0) (in K) - temperature of canonical simulation or temperature to generate velocities.
-
-
ETAWAT (0.8904) - viscosity of water (in centipoises).
-
-
RWAT (1.4) - radius of water molecule (in A)
-
-
SCAL_FRIC=number (real) (0.02) - scaling factor of the friction coefficients.
-
-
SURFAREA - scale friction acting on atoms by atoms' solvent accessible area.
-
-
RESET_FRICMAT=number (integer) (1000) - recalculate friction matrix every RESET_FRICMAT MD steps.
-
-
USAMPL - restraints on q (see reference 5 for meaning) will be imposed (see section . In this case, the next records specify the restraints; these records are placed before the list of temperatures or numbers of trajectories.
-
-
EQ_TIME=number (real) (1.0e4) - time (in mtus; 1 mtu=48.9 fs) after which restraints on q will start to be in force.
If USAMPL has been specified, the following information must be supplied after the main MD input data record (subroutine READ_FRAGMENTS):
Line 1: nset, npair, nfrag_back (number of sets of restraints, number of restrained fragments, number of restrained pairs, number of restrained backbone fragments (in terms of θ and γ angles)
For each set of restraints (1, 2,..., nset):
-
mset(iset) - how many times the set is multiplied.
-
-
wfrag(i,iset), ifrag(1,i,iset), ifrag2(2,i,iset),qfrag(i,iset) - weight of the restraint, first and last residue of the fragment, target q value. This information is repeated through nfrag.
-
-
wpair(i,iset), ipair(1,i,iset), ipair(2,i,iset),qinpair(i,iset) - weight of the restraint, first and second fragment of the pair (according to fragment list), target q value. This information is repeated through npair
-
-
wfrag_back(1,i,iset), wfrag_back(2,i,iset), wfrag_back(3,i,iset), ifrag_back(1,i,iset),ifrag_back(2,i,iset) - weight of the restraints on θ angles, weight on the restraints on γ angles, weight of the restraints on side-chain rotamers, first residue of the fragment, last residue of the fragment. This information is repeated through nfrag_back.
labelsect:input:main:MREMD
(Miced format; subroutine READ_REMDPAR.)
-
NREP (3) - number of replicas in a REMD/MREMD run.
-
-
NSTEX (1000) - number of steps after which exchange is performed in REMD/MREMD runs.
The temperatures in replicas can be specified through
-
-
RETMIN (10.0) - minimum temperature in a REMD/MREMD run,
-
-
RETMAX (1000.0) - maximum temperature in a REMD/MREMD run.
Then the range from retmin to retmax is divided into equal segments and temperature of the replicas assigned accordingly,
or
-
TLIST means that the NREP temperature of the replicas will be input in the next record.
-
-
MLIST numbers of trajectories per each of the NREP temperatures will be specified in the record after the list of temperatures; this specifies a MREMD run.
Important! The number of processors must be exactly equal to the number of trajectories, i.e., NREP for a REMD run or
∑i mlist(i) for a MREMD run.
-
-
SYNC - all trajectories will be synchronized every NSTEX time steps (by default, they are not synchronized).
-
-
TRAJ1FILE - only the master processor outputs coordinates. This feature pertains only to REMD/MREMD jobs and overrides NTWX; coordinates are dumped at every exchange in MREMD.
-
-
REST1FILE - only the master writes the restart file.
-
-
HREMD - Hamiltonian replica exchange flag; not only temperatures but also sets energy-term weights are exchanged between conformations.
-
-
TONLY - run a "fake" HREMD with many sets of energy-term weights in a single run but only temperature exchange.
8.1.8 Energy-term weights
(Data list format; subroutine MOLREAD.)
-
WLONG=number (real) (1.0d0) - common weight of the U(SC-SC) (side-chain side-chain interaction) and U(SC,p) (side-chain peptide-group) term.
-
-
WSCC=number (real) (WLONG) - weight of the U(SC-SC) term.
-
-
WSCP=number (real) (WLONG) weight of the U(SC-p) term.
-
-
WELEC=number (real) (1.0d0) weight of the U(p-p) (peptide-group peptide-group interaction) term.
-
-
WEL_LOC=number (real) (1.0d0) weight of the
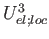 (local-electrostatic cooperativity, third-order) term.
(local-electrostatic cooperativity, third-order) term.
-
-
WCORRH=number (real) (1.0d0) weight of the U(corr) (cooperativity of hydrogen-bonding interactions, fourth-order) term.
-
-
WCORR5=number (real) (0.0d0) - weight of the
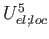 (local-electrostatic cooperativity, 5th order contributions).
(local-electrostatic cooperativity, 5th order contributions).
-
-
WCORR6=number (real) (0.0d0) - weight of the
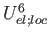 (local-electrostatic cooperativity, 6th order contributions).
(local-electrostatic cooperativity, 6th order contributions).
-
-
WTURN3=number (real) (1.0d0) - weight of the
 (local-electrostatic cooperativity within 3 residue segment, 3rd order contribution).
(local-electrostatic cooperativity within 3 residue segment, 3rd order contribution).
-
-
WTURN4=number (real) (1.0d0) - weight of the
 (local-electrostatic cooperativity within 4 residue segment, 4rd order contributions).
(local-electrostatic cooperativity within 4 residue segment, 4rd order contributions).
-
-
WTURN6=number (real) (1.0d0) - weight of the
 (local-electrostatic cooperativity within 6 residue segment, 6rd order contributions).
(local-electrostatic cooperativity within 6 residue segment, 6rd order contributions).
-
-
WTOR=number (real) (1.0d0) - weight of the torsional term, Utor.
-
-
WANG=number (real) (1.0d0) - weight of the virtual-bond angle bending term, Ub
-
-
WSCLOC=number (real) (1.0d0) - weight of the side-chain rotamer term, USC.
-
-
WSTRAIN=number (real) (1.0d0) - scaling factor of the distance-constrain or disulfide-bond strain energy term.
-
-
SCALSCP=number (real) (1.0d0) - scaling factor of USCp; this is an alternative to specifying WSCP; in this case WSCP will be calculated as WLONG*SCALSCP.
-
-
SCAL14=number (real) (1.0d0) - scaling factor of the 1,4 SC-p interactions.
-
-
CUTOFF (7.0) - cut-off on backbone-electrostatic interactions to compute 4- and higher-order correlations.
-
-
DELT_CORR (0.5) - thickness of the distance range in which the energy is decreased to zero.
The defaults are NOT the recommended values. No "working" default values have been set, because the force field is still under development. The values corresponding to the force fields listed in section 4 are as follows:
CASP3:
WELEC=1.5 WSTRAIN=1.0 WTOR=0.08617 WANG=0.10384 WSCLOC=0.10384 WCORR=1.5 &
WTURN3=0 WTURN4=0 WTURN6=0 WEL_LOC=0 WCORR5=0 WCORR6=0 SCAL14=0.40 SCALSCP=1.0 &
CUTOFF=7.00000 WSCCOR=0.0
ALPHA:
WSC=1.00000 WSCP=0.72364 WELEC=1.10890 WANG=0.68702 WSCLOC=1.79888 &
WTOR=0.30562 WCORRH=1.09616 WCORR5=0.17452 WCORR6=0.36878 WEL_LOC=0.19508 &
WTURN3=0.00000 WTURN4=0.55588 WTURN6=0.11539 CUTOFF=7.00000 WCORR4=0.0000 &
WTORD=0.0 WSCCOR=0.0
BETA:
WSC=1.00000 WSCP=1.10684 WELEC=0.70000 WANG=0.80775 WSCLOC=1.91939 &
WTOR=3.36070 WCORRH=2.50000 WCORR5=0.99949 WCORR6=0.46247 WEL_LOC=2.50000 &
WTURN3=1.80121 WTURN4=4.35377 WTURN6=0.10000 CUTOFF=7.00000 WCORR4=0.00000 &
WSCCOR=0.0
ALPHABETA:
WSC=1.00000 WSCP=1.43178 WELEC=0.41501 WANG=0.37790 WSCLOC=0.12880 &
WTOR=1.98784 WCORRH=2.50526 WCORR5=0.23873 WCORR6=0.76327 WEL_LOC=2.97687 &
WTURN3=0.09261 WTURN4=0.79171 WTURN6=0.01074 CUTOFF=7.00000 WCORR4=0.00000 &
WSCCOR=0.0
CASP5:
WSC=1.00000 WSCP=1.54864 WELEC=0.20016 WANG=1.00572 WSCLOC=0.06764 &
WTOR=1.70537 WTORD=1.24442 WCORRH=0.91583 WCORR5=0.00607 WCORR6=0.02316 &
WEL_LOC=1.51083 WTURN3=2.00764 WTURN4=0.05345 WTURN6=0.05282 WSCCOR=0.0 &
CUTOFF=7.00000 WCORR4=0.00000 WSCCOR=0.0
3P:
WSC=1.00000 WSCP=2.85111 WELEC=0.36281 WANG=3.95152 WSCLOC=0.15244 &
WTOR=3.00008 WTORD=2.89863 WCORRH=1.91423 WCORR5=0.00000 WCORR6=0.00000 &
WEL_LOC=1.72128 WTURN3=2.99827 WTURN4=0.59174 WTURN6=0.00000 &
CUTOFF=7.00000 WCORR4=0.00000 WSCCOR=0.0
4P:
WSC=1.00000 WSCP=2.73684 WELEC=0.06833 WANG=4.15526 WSCLOC=0.16761 &
WTOR=2.99546 WTORD=2.89720 WCORRH=1.98989 WCORR5=0.00000 WCORR6=0.00000 &
WEL_LOC=1.60072 WTURN3=2.36351 WTURN4=1.34051 WTURN6=0.00000 &
CUTOFF=7.00000 WCORR4=0.00000 WSCCOR=0.0
GAB:
WLONG=1.35279 WSCP=1.59304 WELEC=0.71534 WBOND=1.00000 WANG=1.13873 &
WSCLOC=0.16258 WTOR=1.98599 WTORD=1.57069 WCORRH=0.42887 WCORR5=0.00000 &
WCORR6=0.00000 WEL_LOC=0.16036 WTURN3=1.68722 WTURN4=0.66230 WTURN6=0.00000 &
WVDWPP=0.11371 WHPB=1.00000 &
CUTOFF=7.00000 WCORR4=0.00000
E0G:
WLONG=1.70905 WSCP=2.18310 WELEC=1.06684 WBOND=1.00000 WANG=1.17536 &
WSCLOC=0.22070 WTOR=2.65798 WTORD=2.00646 WCORRH=0.23541 WCORR5=0.00000 &
WCORR6=0.00000 WEL_LOC=0.42789 WTURN3=1.68126 WTURN4=0.75080 WTURN6=0.00000 &
WVDWPP=0.27044 WHPB=1.00000 WSCP14=0.00000 &
CUTOFF=7.00000 WCORR4=0.00000
E0LL2Y:
WLONG=1.00000 WSCP=1.23315 WELEC=0.84476 WBOND=1.00000 WANG=0.62954 &
WSCLOC=0.10554 WTOR=1.84316 WTORD=1.26571 WCORRH=0.19212 WCORR5=0.00000 &
WCORR6=0.00000 WEL_LOC=0.37357 WTURN3=1.40323 WTURN4=0.64673 WTURN6=0.00000 &
WVDWPP=0.23173 WHPB=1.00000 WSCCOR=0.0 &
CUTOFF=7.00000 WCORR4=0.00000
8.1.9 Input and/or reference PDB file name
(Text format; subroutine MOLREAD.)
If PDBSTART or PDBREF was specified in the control card, this line contains the PDB file name. Trailing slashes to specify the full path are permitted. The file name can contain up to 64 characters.
8.1.10 Amino-acid sequence
(Mixed format.)
This data appears, if PDBSTART was not specified, otherwise must not be present because the sequence would be taken from the PDB file. The first line contains the number of amino-acid residues, including the end groups (free format), the next lines contain the sequence in 20(1X,A3) format for the three-letter or 80A1 format for the one-letter code. There are two types of end-groups: Gly (three-letter code) or G (one-letter code), if an end group contains a full peptide bond (e.g., the acetyl N-terminal group or the carboxyamide C-terminal group) and D (in the three-letter code) or X (in the one-letter code), if the end group does not contain a peptide group (e.g., the NH2 N-terminal end group or the COOH C-terminal end group). (Note the Gly or G also denotes the regular glycine residue, if found in the middle of a chain). In the second case the end group is considered as a "dummy" group and serves only to define the first (last) virtual-bond dihedral angle γ for the first (last) full amino-acid residue.
Consider, for example, the Ac-Ala(19)-NHMe polypeptide. The three-letter code input will look like this:
21
Gly Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala
Gly
And the one-letter code input will be:
21
GAAAAAAAAAAAAAAAAAAAG
If the sequence is changed to NH3(+)-Ala(19)-COO(-), the inputs will look like this:
21
D Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala
D
and
21
XAAAAAAAAAAAAAAAAAAAX
The sequence input is case-insensitive, because the present version of UNRES considers each amino-acid residue as an L-residue (there are no torsional parameters for the combinations of the D- and L-residues yet). Furthermore, each peptide group is considered as a trans group.
If the version of UNRES has multi-chain capacity, placing a dummy residue inside the sequence indicates start of a new chain. For example, a system composed of two Ala(10) chains can be specified as follows (3-letter code):
23
D Ala Ala Ala Ala Ala Ala Ala Ala Ala Ala D Ala Ala Ala Ala Ala Ala Ala Ala
Ala Ala D
or (1-letter code)
23
XAAAAAAAAAAXAAAAAAAAAAX
8.1.11 Disulfide-bridge information
(Free format; subroutine READ_BRIDGE.)
1st line:
NS,(ISS(i),i=1,NS)
-
NS - the number of half-cystines (required even if no half-cystines are present).
-
-
ISS(i) - the position of ith half-cystine in the sequence (starting from the N-terminal end group)
Next line(s) (present only, if ns > 0 and must not appear otherwise):
NSS,(IHPB(i),JHPB(i),i=1,NSS)
-
NSS - the number of disulfide bridges; must not be greater than NS/2.
-
-
IHPB(i),JHPB(i) - the cystine residue forming the ith bridge.
The program will check, whether the residues specified in the ISS list are cystines and terminate with error, if any of them is not. The program also checks, if the numbers from the IHPB and the JHPB lists have appeared in the ISS list.
8.1.12 Dihedral-angle restraint data
(Free format; subroutine MOLREAD.)
This set of data specifies the harmonic constraints (if any) imposed on selected virtual-bond dihedral angles γ.
1st line:
-
NDIH_CONSTR - the number of restrained γ angles (required even if no restrains are applied).
2nd line (present only, if NDIH_CONSTR > 0; must not appear otherwise): FTORS - the force constant expressed in kcal/(mol*rad**2)
next NDIH_CONSTR lines (present only, if NDIH_CONSTR > 0):
IDIH_CONSTR(i),PHI0(i),DRANGE(i)
-
-
IDIH_CONSTR(i) - the number of ith restrained γ angle. The angles are numbered after the LAST α-carbons. Thus, the first "real" angle has number 4 and it corresponds to the rotation about the CA(2)-CA(3) virtual-bond axis and the last angle has the number NRES and corresponds to the rotation about the CA(NRES-2)-CA(NRES-1) virtual-bond axis.
-
-
PHI0(i) - the "center" of the restraint (expressed in degrees).
-
-
DRANGE(i) - the "flat well" range of the restraint (in degrees).
The restraint energy for the ith restrained angle is expressed as:
Applying dihedral-angle constraints also implies that for ith constrained γ angle the sampling be carried out from the [PHI0(i)-DRANGE(i)..PHI0(i)+DRANGE(i)] interval and not from the [−π…π] interval, if random conformations are generated. If only this and not restrained minimization is required, just set FTORS to 0.
8.1.13 Distance restraints
(Mixed format; subroutine READ_DIST_CONSTR.)
Restraints are imposed on C
α…Cα SC…SC distances (Cβ…Cβ).
-
NDIST=number (integer) (0) - number of restraints on specific distances.
-
-
NFRAG=number (integer) (0) - number of distance-restrained protein segments.
-
-
NPAIR=number (integer) (0) - number of distance-restrained pairs of segments. Specifying NPAIR requires specification of segments.
-
-
IFRAG=start(1),end(1),start(2),end(2)...start(NFRAG),end(NFRAG) (integers) - First and last residues of the distance restrained segments.
-
-
WFRAG=w(1),w(2),...,w(NFRAG) (reals) - force constants or bases for force constant calculation corresponding to fragment restraints.
-
-
IPAIR=start(1),end(1),start(2),end(2),...,start(NPAIR),end(NPAIR) (integers) - numbers of segments (consecutive numbers of start or end pairs in IFRAG specification), the distances between which will be restrained.
-
-
WPAIR=w(1),w(2),...,w(NFRAG) (reals) - force constants or bases for force constant calculation corresponding to pair restraints.
-
DIST_CUT=number (real) (5.0) - the cut-off distance in angstroms for force- constant calculations.
The force constants within fragments/between pairs of fragments are calculated depending on the value of DIST_CONSTR described in section 5.1:
-
-
1 - all force constants are equal to the respective entries of WFRAG/WPAIR
-
-
2 - the force constants are equal to the respective entries of WFRAG/WPAIR when the distance between the Cα atoms in the reference structure ≤ D_CUT, 0 otherwise.
-
-
3 - the force constants are calculated from the formula:
-
-
![$ k(C^\alpha_j,C^\alpha_k)=W\times\exp{-[d(C^\alpha_j,C^\alpha_k)/DIST\_CUT)]^2/2}$](/sites/unres/files/v3/img/img34.png)
-
where 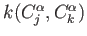 is the force constant between the respective Cα atoms,
is the force constant between the respective Cα atoms, 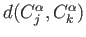 is the distance between these Cα atoms in the reference structure, and W is the basis for force-constant calculation (see above).
is the distance between these Cα atoms in the reference structure, and W is the basis for force-constant calculation (see above).
The above restraints are harmonic resatraints of the form
where di is the distance in the calculated structure and 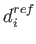 is the respective distance in the reference (PDB) structure. The reference structure is required.
is the respective distance in the reference (PDB) structure. The reference structure is required.
If NDIST > 0, the restraints on specific distance are input explicitly (no reference structure is requires). The restraints are quartic restraints of a similar form as that in section 8.1.12 but with angles replaced with distances.
ihpb(i), jhpb(i), dhpb(i), dhpb1(i), ibecarb(i), forcon(i), i=1,NDIST
-
-
ihpb(i) and jhpb(i) are the numbers of the residues the distance between the Cα atoms of which will be distance restrained,
-
-
dhpb(i) and dhpb1(i) are the lower and upper distance-restraint,
-
-
ibecarc(i) is the restraint-type flag; ibecarb(i)==0 indicates that the restraints are imposed on the
Cα … Cα distances; otherwise restraints on the SC…SC distances are imposed,
-
-
forcon(i) is the respective force constant.
8.1.14 Internal coordinates of the reference structure
(Free format; subroutine READ_ANGLES.)
This part of the data is present, if REFSTR, but not PDBREF was specified, otherwise must not appear. It contains the following group of variables:
-
(THETA(i),i=3,NRES) - the virtual-bond valence angles THETA.
-
-
(PHI(i),i=4,NRES) - the virtual-bond dihedral angles GAMMA.
-
-
(ALPH(i),i=2,NRES-1) - the ALPHA polar angles of consecutive side chains.
-
-
(OMEG(i),i=2,NRES-1) - the BETA polar angles of consecutive side chains.
ALPHA(i) and OMEG(i) correspond to the side chain attached to CA(i). THETA(i) is the CA(i-2)-CA(i-1)-CA(i) virtual-bond angle and PHI(i) is the CA(i-3)-CA(i-2)-CA(i-1)-CA(i) virtual-bond dihedral angle γ.
8.1.15 Internal coordinates of the initial conformation
(Free format; subroutine READ_ANGLES.)
This part of the data is present, if RAND_CONF, MULTCONF, THREAD, or PDBSTART were not specified, otherwise must not appear. This input is as in section 10.
8.1.15.1 File name with internal coordinates of the conformations to be processed
(Text format; subroutine MOLREAD.)
This data is present only, if MULTCONF was specified. It contains the name of the file with the internal coordinates. Up to 64 characters are allowed. The structure of the file is that of the *.int file produced by UNRES/CSA. See section "The structure of the INT files" for details.
8.1.16 Control data for energy map construction
(Data list format; subroutine MAP_READ.)
These data lists appear, if NMAP=n was specified, where n is the number of variables that will be grid-searched. One list is per one variable or a group of variables set equal (see below):
-
PHI - the variable is a virtual-bond dihedral angle γ.
-
-
THE - the variable is a virtual-bond angle θ.
-
-
ALP - the variable is a side-chain polar angle α.
-
-
OME - the variable is a side-chain polar angle β.
-
-
RES1=number (integer)
-
-
RES2=number (integer)
The range of residues for which the values will be set; all these variables will be set at the same value. It is required that RES2 > RES1.
-
FROM=angle (real)
-
-
TO=angle (real)
Lower and upper limit of scanning in grid search (in degrees)
-
-
NSTEP=number (integer)
Number of steps in scanning along this variable/group of variables.
8.2 Input coordinate files
(Text format; subroutine MOLREAD.)
At present, geometry can be input either from the external files in the PDB format (with the PDBSTART option) or multiple conformations can be read as virtual-bond-valence and virtual-bond dihedral angles when the MULTCONF option is used (the latter, however, implies using standard virtual-bond lengths as initial values). The structure of internal-coordinate files is the same as that of output internal-coordinate files described in section 9.1.1.
8.3 Other input files
CSA parameters can optionally be read in free format from file INPUT.CSA.in (see section 8.1.4). When a CSA run is restarted, the CSA-specific output files also serve as input files. INPUT is the prefix of input and output files as explained in section 6.
Restart files for MD and REMD simulations. They are read when the keyword RESTART appears on the MD/REMD data group (section 8.1.6).
9 OUTPUT FILES
UNRES "main" output files (INPUT.out_${POT}[processor]) are log files from a run. They contain the information of the molecule, force field, calculation type, control parameters, etc.; however, not the structures produced during the run or their energies except single-point energy evaluation and minimization-related runs. The structural information is included in coordinate files (*.int, *.x, *.pdb, *.mol2, *.cx) and statistics files (*.stat), respectively; these files are further processed by other programs (WHAM, CLUSTER) or can be viewed by molecular viewers (pdb or mol2 files).
9.1 Coordinate files
9.1.1 The internal coordinate (INT) file
This file contains the internal coordinates of the conformations produced by UNRES in non-MD runs. The virtual-bond lengths are assumed constant so only the angular variables are provided.
IT,ENER,NSS,(IHPB(I),JHPB(I),I=1,NSS)
(I5,F12.5,I2,9(1X,2I3))
-
-
IT - the number of the conformation.
-
-
ENER - total energy.
-
-
NSS - the number of disulfide bridges.
-
-
(IHPB(I),JHPB(I),I=1,NSS) - the positions of the pairs of half-cystines . forming the bridges. If NSS > 99, the remaining pairs are written in the following lines in the (3X,11(1X,2I3)) format.
(THETA(I),I=3,NRES)
(8F10.4)
The virtual-bond angles THETA (in degrees)
(PHI(I),I=4,NRES)
(8F10.4)
The virtual-bond dihedral angles GAMMA (in degrees)
(ALPH(I),I=2,NRES-1)
(OMEG(I),I=2,NRES-1)
(8F10.4)
The polar angles ALPHA and BETA of the side-chain centers (in degrees).
9.1.2 The plain Cartesian coordinate (X) files
(Subroutine CARTOUT.)
This file contains the Cartesian coordinates of the α-carbon and side-chain-center coordinates. All conformations from an MD/MREMD trajectory are collated to a single file. The structure of each conformation's record is as follows:
1st line: time, potE, uconst, t_bath,nss, (ihpb(j), jhpb(j), j=1,nss), nrestr, (qfrag(i), i=1,nfrag), (qpair(i), i=1,npair), (utheta(i), ugamma(i), uscdiff(i), i=1,nfrag_back)
-
-
time: MD time (in "molecular time units" 1 mtu = 4.89 fs),
-
-
potE: potential energy,
-
-
uconst: restraint energy corresponding to restraints on Q and backbone geometry, (see section 8.1.6),
-
-
t_bath: thermostat temperature,
-
-
nss: number of disulfide bonds,
-
-
ihpb(j), jhpb(j): the numbers of linked cystines for jth disulfide bond,
-
-
nrestr: number of restraints on q and local geometry,
-
-
qfrag(i): q value for ith fragment,
-
-
qpair(i): q value for ith pair,
-
-
utheta(i): sum of squares of the differences between the theta angles of the current conformation from those of the experimental conformation,
-
-
ugamma(i): sum of squares of the differences beaten the gamma angles of the current conformation from those of the experimental conformation,
-
-
uscdiff(i): sum of squares of the differences between the Cartesian difference of the unit vector of the Cα-SC axis of the current conformation from those of the experimental conformation.
Next lines: Cartesian coordinates of the Cα atoms (including dummy atoms) (sequentially, 10 coordinates per line) Next lines: Cartesian coordinates of the SC atoms (including glycines and dummy atoms) (sequentially, 10 coordinates per line)
9.1.3 The compressed Cartesian coordinate (CX) files
These files are compressed binary files (extension cx). For each conformation, the items are written in the same order as specified in section 9.1.3. For MREMD runs, if TRAJ1FILE is specified on MREMD record (see section 8.1.6), snapshots from all trajectories are written every time the coordinates are dumped. Thus, the file contains snapshot 1 from trajectory 1, ..., snapshot 1 from trajectory M, snapshot 2 from trajectory 1, ..., etc.
The compressed cx files can be converted to pdb file by using the xdrf2pdb auxiliary program (single trajectory files) or xdrf2pdb-m program (multiple trajectory files from MREMD runs generated by using the TRAJ1FILE option). The multiple-trajectory cx files are also input files for the auxiliary WHAM program.
9.1.4 The Brookhaven Protein Data Bank format (PDB) files
(Subroutine PDBOUT.)
These files are written in PDB standard (see. e.g., ftp://ftp.wwpdb.org/pub/pdb/doc/format_descriptions/Format_v33_Letter.pdf). The REMARK, ATOM, SSBOND, HELIX, SHEET, CONECT, TER, and ENDMDL are used. The Cα (marked CA) and SC (marked CB) coordinates are output. The CONECT records specify the
Cα… Cα and Cα … SC virtual bonds. Secondary structure is detected based on peptide-group contacts, as specified in ref 12. Dummy residues are omitted from the output. If the program has multiple-chain function, the presence of a dummy residue in a sequence starts a new chain, which is assigned the next alphabet letter as ID, and residue numbering is started over.
9.1.5 The SYBYLL (MOL2) files
See the description of mol2 format (e.g., http://tripos.com/data/support/mol2.pdfhttp://tripos.com/data/support/mo.... Similar remarks apply as for the PDB format (section 9.1.4).
This file contains a short summary of the quantities characterizing the conformations produced by UNRES/CSA. It is created for MULTCONF and MCM.
NOUT,EVDW,EVDW2,EVDW1+EES,ECORR,EBE,ESCLOC,ETORS,ETOT,RMS,FRAC
(I5,9(1PE14.5))
-
NOUT - the number of the conformations
-
-
EVDW,EVDW2,EVDW1+EES,ECORR,EBE,ESCLOC,ETORS - energy components
-
-
ETOT - total energy
-
-
RMS - RMS deviation from the reference structure (if REFSTR was specified)
-
-
FRAC - fraction of side chain - side chain contacts of the reference structure present in this conformation (if REFSTR was specified)
9.2.2 MD and MREMD runs
Each line of the stat file generated by MD/MREMD runs contains the following items in sequence:
-
-
step - the number of the MD step
-
-
time - time [unit is MTU (molecular time unit) equal to 48.9 fs]
-
-
Ekin - kinetic energy [kcal/mol]
-
-
Epot - potential energy [kcal/mol]
-
-
Etot - total energy (Ekin+Epot)
-
-
H-H0 - the difference between the cureent and initial extended Hamiltionian in Nose-Hoover or Nose-Poincare runs; not present for other thermostats.
-
-
RMSD - root mean square deviation from the reference structure (only in REFSTR has been specified) itemdamax - maximum change of acceleration between two MD steps
-
-
fracn - fraction of native side-chain concacts (very crude, based on SC-SC distance only)
-
-
fracnn - fraction of non-native side-chain contacts
-
-
co - contact order
-
-
temp - actual temperature [K]
-
-
T0 - initial (microcanonical runs) or thermostat (other run types) temperature [K]
-
-
Rgyr - radius of gyration based on Cα coordinates [A]
-
-
proc - in MREMD runs the number of the processor (the number of the trajectory less 1); not present for other runs.
For an USAMPL run, the following items follow the above list:
-
-
iset - the number of the restraint set
-
-
uconst - restraint energy pertaining to q-values
-
-
uconst_back - restraint energy pertaining to virtual-backbone restraints
-
-
(qfrag(i),i=1,nfrag) - q values of the specified fragments
-
-
(qpair(ii2),ii2=1,npair) - q values of the specified pairs of fragments
-
-
(utheta(i),ugamma(i),uscdiff(i),i=1,nfrag_back) - virtual-backbone and side-chain-rotamer restraint energies of the fragments specified
If PRINT_COMPON has been specified, the energy components are printed after the items described above.
9.3 CSA-specific output files
There are several output files from the CSA routine: INPUT.CSA.seed, INPUT.CSA.history, INPUT.CSA.bank, INPUT.CSA.bank1, INPUT.CSA.rbank INPUT.CSA.alpha, INPUT.CSA.alpha1.
The most informative outfile is INPUT.CSA.history. This file first write down the parameters in INPUT.CSA.csa file. Later it shows the energies of random minimized conformations in its generation. After sorting the First_bank in energy (ascending order), the energies of the First_bank is re-written here. After this the output looks like:
1 0 100 6048.2 1 100-224.124-114.346 202607 100 100
1 0 700 5882.6 2 29-235.019-203.556 1130308 100 100
1 0 1300 5721.5 2 18-242.245-212.138 2028008 100 100
1 0 1900 5564.8 13 54-245.185-218.087 2897988 98 100
1 0 2500 5412.4 13 61-246.214-222.068 3706478 97 100
1 0 3100 5264.2 13 89-248.715-224.939 4514196 96 100
Each line is written between each iteration (just after selection of seed conformations) containing following data: jlee,icycle,nstep,cutdif,ibmin,ibmax,ebmin,ebmax,nft,iuse,nbank ibmin and ibmax lists the index of bank conformations corresponding to the lowest and highest energies with ebmin and ebmax. nft is the total number of function evaluations so far. iuse is the total number of conformations which have not been used as seeds prior to calling subroutine select_is which select seeds.
Therefore, in the example shown above, one notes that so far 3100 minimizations has been performed corresponding to the total of 4514196 function evaluations. The lowest and highest energy in the Bank is -248.715 (#13) and -224.939 (#89), respectively. The number of conformations already used as seeds (not including those selected as seeds in this iteration) so far is 4 (100-96).
The files INPUT.CSA.bank and INPUT.CSA.rbank contains data of Bank and First_bank. For more information on these look subroutines write_bank and write_rbank. The file INPUT.CSA.bank is overwritten between each iteration whereas Bank is accumulated in INPUT.CSA.bank1 (not for every iteration but as specified in the subroutine together.f).
The file INPUT.CSA.seed lists the index of the seed conformations with their energies. Files INPUT.CSA.alpha, INPUT.CSA.alpha1 are written only once at the beginning of the CSA run. These files contain some arrays used in CSA procedure.
10 TECHNICAL SUPPORT CONTACT INFORMATION
Dr. Adam Liwo
Faculty of Chemistry, University of Gdansk
ul. Wita Stwosza 63, 80-308 Gdansk Poland.
phone: +48 58 523 5124
fax: +48 58 523 5012
e-mail: adam(at)sun1.chem.univ.gda.pl
Dr. Cezary Czaplewski
Faculty of Chemistry, University of Gdansk
ul. Wita Stwosza 63, 80-308 Gdansk Poland.
phone: +48 58 523 5126
fax: +48 58 523 5012
e-mail: cezary.czaplewski(at)ug.edu.pl
Dr. Adam Sieradzan
Faculty of Chemistry, University of Gdansk
ul. Wita Stwosza 63, 80-308 Gdansk Poland.
phone: +48 58 523 5124
fax: +48 58 523 5012
e-mail: adasko(at)sun1.chem.univ.gda.pl
Dr. Stanislaw Oldziej
Intercollegiate Faculty of Biotechnology
University of Gdansk, Medical University of Gdansk
ul. Kladki 22, 80-922 Gdansk, Poland
phone: +48 58 523 5361
fax: +48 58 523 5472
e-mail: stan(at)biotech.ug.edu.pl
Dr. Jooyoung Lee
Korea Institute for Advanced Study
207-43 Cheongnyangni 2-dong, Dongdaemun-gu,
Seoul 130-722, Korea
phone: +82-2-958-3890
fax: +82-2-958-3731
email: jlee(at)kias.re.kr
Prepared by Adam Liwo and Jooyoung Lee, 7/17/99
Revised by Cezary Czaplewski 1/4/01
Revised by Cezary Czaplewski and Adam Liwo 8/26/03
Revised by Cezary Czaplewski and Adam Liwo 11/26/11
Revised by Adam Liwo 02/19/12
LaTeX version by Adam Liwo 09/25/12
Revised by Adam Liwo 12/04/14
This document is available in the PDF format also.