
UNRES web server
now available:UNRES web server
UNRES (from UNited RESidue) is a reduced model of polypeptide chains, developed in the groups of Harold A. Scheraga, Cornell University, USA and Adam Liwo, University of Gdansk, Poland. Initially, it was designed for physics-based predictions of protein structure through global optimization of an effective potential-energy function of polypeptide chains plus solvent. Later, it was developed to include molecular dynamics algorithm and generalized-ensemble sampling algorithms of which multiplexed replica exchange molecular dynamics (MREMD) turned out to be the most efficient.
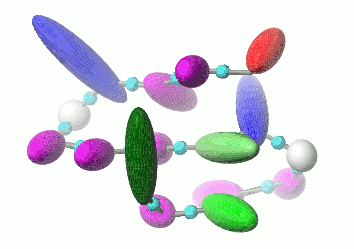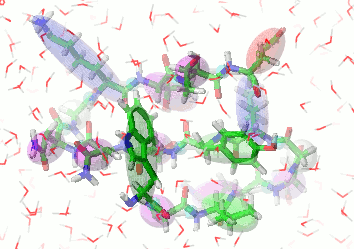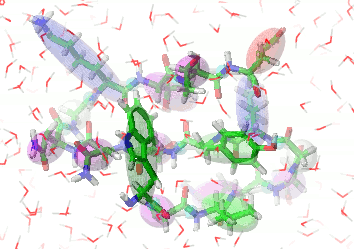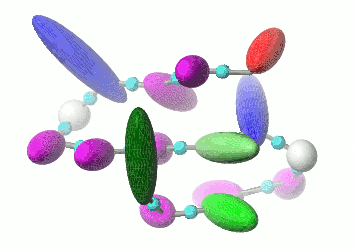 UNRES is a highly reduced protein model; only two interaction sites: united side chain and united peptide group per residue are present. Owing to this reduction, it offers ~1000-4000-fold speed up in molecular dynamics simulations compared to all-atom approaches. With recently introduced parallelization of energy and force evaluation, it enables us to perform ab initio folding simulations of 200-residue proteins in hours and simulations of large biologically inportant conformational changes in large proteins (e.g., molecular chaperones) in days of wall-clock time.
UNRES is a highly reduced protein model; only two interaction sites: united side chain and united peptide group per residue are present. Owing to this reduction, it offers ~1000-4000-fold speed up in molecular dynamics simulations compared to all-atom approaches. With recently introduced parallelization of energy and force evaluation, it enables us to perform ab initio folding simulations of 200-residue proteins in hours and simulations of large biologically inportant conformational changes in large proteins (e.g., molecular chaperones) in days of wall-clock time.
The UNRES force field has been developed on a solid statistical-mechanical basis, by expanding the potential of mean force of a system containing polypeptide chain(s) in water into cluster-cumulant series and parameterization of the terms of the series (factors) based on simple model systems. Therefore, even though no knowledge-based information is used in simulations (from homology modeling, loop and contact prediction, etc.), the force field, in its present version can be used in ab initio folding simulations and ab initio prediction of protein structures to predict the folds of fragments with 50-200 residues in length.
UNRES fared very well in the CASP3 blind-prediction exercise and continued to yield good predictions in subsequent exercises. Present work is directed at improvement of the representation of the local and side chain-side chain interactions as well as force-field optimization to increase the accuracy.
UNRES is a very good system to study protein folding by molecular simulations, which enables us running hundreds of folding trajectories simultaneously or to study thermodynamics of protein folding by using replica-exchange simulations. It also enable us to run large-scale simulations aimed at discerning functionally important motions of large proteins. Examples of its past and present biological applications include studies of amyloid formations, mechanism of signaling, and action of molecular chaperones.
This site contains the full version of the UNRES package for all applications and with all auxiliary programs. The package will be updated shortly to include more examples and automatic installation procedures.
For detailed description refer to the DOCS section. Download the package or its parts as gzipped tarfile(s) from the DOWNLOADS section.