WHAM
(Weighted Histogram Analysis Method)
Processing results of UNRES/MREMD simulations
Laboratory of Molecular Modeling
Faculty of Chemistry
University of Gdansk
Wita Stwosza 63
80-308 Gdansk, Poland
Scheraga Group
Baker Laboratory of Chemistry
and Chemical Biology
Cornell University
Ithaca, NY 14853-1301, USA
December 4, 2014
1 LICENSE TERMS
-
This software is provided free of charge to academic users, subject to the condition that no part of it be sold or used otherwise for commercial purposes, including, but not limited to its incorporation into commercial software packages, without written consent from the authors. For permission contact Prof. H. A. Scheraga, Cornell University.
-
This software package is provided on an "as is" basis. We in no way warrant either this software or results it may produce.
-
Reports or publications using this software package must contain an acknowledgment to the authors and the NIH Resource in the form commonly used in academic research.
2 REFERENCES
Citing the following references in your work that makes use of the WHAM software is gratefully acknowledged:
-
[1]
-
S. Kumar, D. Bouzida, R.H. Swendsen, P.A. Kollman, J.M. Rosenberg. The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem., 1992, 13, 1011-1021.
-
[2]
-
A. Liwo, M. Khalili, C. Czaplewski, S. Kalinowski, S. Oldziej, K. Wachucik, H.A. Scheraga. Modification and optimization of the united-residue (UNRES) potential energy function for canonical simulations. I. Temperature dependence of the effective energy function and tests of the optimization method with single training proteins. J. Phys. Chem. B, 2007, 111, 260-285.
-
[3]
-
S. Oldziej, A. Liwo, C. Czaplewski, J. Pillardy, H.A. Scheraga. Optimization of the UNRES force field by hierarchical design of the potential-energy landscape. 2. Off-lattice tests of the method with single proteins. J. Phys. Chem. B, 2004, 108, 16934-16949.
3 FUNCTIONS OF THE PROGRAM
The program processes the results of replica exchange (REMD) or multiplexed replica exchange molecular dynamics (MREMD) simulations with UNRES to compute the probabilities of the obtained conformations to occur at particular temperatures. The program is based on the variant of the weighted histogram analysis (WHAM) method [1] described in ref [2].
The program outputs the following information:
-
(a)
-
Temperature profiles of thermodynamic and structural ensemble-averaged quantities.
-
(b)
-
Histograms of native-likeness measure q (defined by eqs 8-11 of ref [[2]]).
-
(c)
-
Optionally the most probable conformations at REMD temperatures.
-
(d)
-
Optionally the coordinates with information to compute probabilities for the conformations to occur at any temperature.
The program takes usually UNRES compressed coordinate files (cx files) from MREMD obtained by using the TRAJ1FILE option. The user can request to partition the whole run into equal slices (or windows), each starting from, say, snapshot n (for each trajectory) and ending at snapshot n+1. Alternatively, the UNRES Cartesian coordinate (x files) can be input; however, they must contain only the analyzed portion of the trajectories; they are usually prepared from single trajectories by using xdrf2x.
Two versions of the program are provided:
-
(a)
-
Canonical version which treats single polypeptide chains; the source code is in WHAM/src directory.
-
(b)
-
Version for oligomeric proteins; multiple chains are handled by inserting dummy residues in the sequence; the source code is in WHAM/src-M directory.
4 INSTALLATION
It is recommended to use Cmake to install the entire package; see the Installation Guide for instructions.
Step-by-step installation without Cmake is also possible; please follow section 4 of Installation Guide for general
information.
Customize Makefile to your system. See section 7 of the description of UNRES for compiler flags that are used to created executables for a particular force field. There are already several Makefiles prepared for various systems and force fields.
Run make in the WHAM/src directory WHAM/src-M directory for multichain version. Make sure that MPI is installed on your system; the present program runs only in parallel mode.
5 RUNNING THE PROGRAM
The program requires a parallel system to run. Depending on system, either the wham.csh C-shell script (in WHAM/bin directory) can be started using mpirun or the binary in the C-shell script must be executed through mpirun. See the wham.csh C-shell script and section 6 for the files processed by the program.
6 INPUT AND OUTPUT FILES
6.1 Summary of the files
The C-shell script wham.csh is used to run the program (see the WHAM/bin directory). The data files that the script needs are mostly the same as for UNRES (see section 6 of UNRES description). In addition, the environmental variable CONTFUN specifies the method to assess whether two side chains are at contact; if CONTFUN=GB, the criterion defined by eq 8 of ref 4 is used to assess whether two side chains are at contact. Also, the parameter files from the C-shell scripts are overridden if the data from Hamiltonian MREMD are processed; if so, the parameter files are defined in the main input file.
The main input file must have inp extension. If it is INPUT.inp, the output files are as follows:
-
INPUT.out_POTxxx - output files from different processors (INPUT.out_000 is the main output file). POT is the identifier of the sidechain-sidechain potential.
-
-
INPUT_POT_GB_xxx.stat or INPUT_POT_slice_YYXXX.stat - the summary conformation-classification file from processor xxx (each processor handles part of conformations); the second occurs if the run is partitioned into slices.
-
-
INPUT.thermal or INPUT_slice_yy.thermal - thermodynamic functions and temperature profiles of the ensemble averages (the second form if the run is partitioned into slices).
-
-
INPUT_T_xxx.pdb or INPUT_slice_yy_T_xxx.pdb - top conformations the number of these conformations is selected by the user) in PDB format.
-
-
INPUT.cx - the compressed UNRES coordinate file with information to compute the probability of a given conformation at any temperature.
-
-
INPUT.hist, INPUT_slice_xx.hist, INPUT_par_yy.hist, INPUT_par_yy_slice_zz.x - histograms of q at MREMD temperatures.
-
-
INPUT.ent, INPUT_slice_xx.ent, INPUT_par_yy.ent, INPUT_par_yy_slice_xx.ent - the histogram(s) of energy density.
-
-
INPUT.rmsrgy, INPUT_par_yy.rmsrgy, INPUT_slice_xx.rmsrgy or INPUT_par_yy_slice_xx.rmsrgy - the 2D histogram(s) of rmsd from the experimental structure and radius of gyration.
6.2 Main input file
This file has the same structure as the UNRES input file; most of the data are input in a keyword-based form (see section 7.1 of UNRES description). The data are grouped into records, referred to as lines. Each record, except for the records that are input in non-keyword based form, can be continued by placing an ampersand (&) in column 80. Such a format is referred to as the data list format.
In the following description, the default values are given in parentheses.
6.2.1 General data (data list format)
-
-
N_ENE (N_ENE_MAX) - the number of energy components.
-
-
SYM (1) - number of chains with same sequence (for oligomeric proteins only).
-
-
HAMIL_REP - if present, Hamiltonian process the results of replica exchange runs (replicas with different parameters of the energy function).
-
-
NPARMSET (1) - number of energy parameter sets ( >1 only for Hamiltonian replica exchange simulations).
-
-
SEPARATE_PARSET - if present, HREMD was run in a mode such that only temperature but not energy-function parameters was exchanged.
-
-
IPARMPRINT (1) - number of parameter set with which to construct conformational ensembles; important only when HREMD runs are processed.
-
-
ENE_ONLY - if present, only conformational energies will be calculated and printed; no WHAM iteration.
-
-
EINICHECK (2) - >0 compare the conformational energies against those stored in the coordinate file(s); 1: compare but print only a warning message if different; 2: compare and terminate the program if different; 0: don't compare.
-
MAXIT (5000) - maximum number of iterations in solving WHAM equations.
-
ISAMPL (1) - input conformation sampling frequency (e.g., if ISAMPL=5, only each 5th conformation will be read).
-
-
NSLICE (1) - number of "slices" or "windows" into which each trajectory will be partitioned; each slice will be analyzed independently.
-
-
FIMIN (0.001) - maximum average difference between window free energies between the current and the previous iteration.
-
-
ENSEMBLES (0) - number of conformations (ranked according to probabilities) to be output to PDB file at each MREMD temperature; 0 means that no conformations will be output. Non-zero values should not be used when NSLICE >1.
-
-
CLASSIFY - if present, each conformation will be assigned a class, according to the scheme described in ref [3].
-
-
DELTA (0.01) - one dimension bin size of the histogram in q.
-
-
DELTRMS (0.05) - rms dimension bin size in rms-radius of gyration histograms.
-
-
DELTRGY (0.05) - radius of gyration bin size in rms-radius of gyration histograms.
-
-
NQ (1) - number of q's (can be for entire molecule, fragments, and pairs of fragments).
-
-
CXFILE - produce the compressed coordinate file with information necessary to compute the probabilities of conformations at any temperature.
-
-
HISTOUT - if present, the histograms of q at MREMD temperatures are constructed and printed to main output file.
-
-
HISTFILE - if present, the histograms are also printed to separate files.
-
-
ENTFILE - if present, histogram of density of states (entropy) is constructed and printed.
-
-
RMSRGYMAP - if present, 2D histograms of radius of rmsd and radius of gyration at MREMD temperatures are constructed and printed.
-
-
WITH_DIHED_CONSTR - if present, dihedral-angle restraints were imposed in the processed MREMD simulations.
-
-
RESCALE (1) - Choice of the type of temperature dependence of the force field.
-
-
>0 - no temperature dependence.
-
1 - homographic dependence (not implemented yet with any force field).
-
2 - hyperbolic tangent dependence [2].
6.2.2 Molecule data
6.2.2.1 General information
-
-
SCAL14 (0.4) - scale factor of backbone-electrostatic 1,4-interactions.
-
SCALSCP (1.0) - scale factor of SC-p interactions.
-
-
CUTOFF (7.0) - cut-off on backbone-electrostatic interactions to compute 4- and higher-order correlations.
-
-
DELT_CORR (0.5) - thickness of the distance range in which the energy is decreased to zero.
-
-
ONE_LETTER - if present, the sequence is to be read in 1-letter code, otherwise 3-letter code.
6.2.2.2 Sequence information
1st record (keyword-based input):
NRES - number of residues, including the UNRES dummy terminal residues, if present
Next records: amino-acid sequence
3-letter code: Sequence is input in format 20(1X,A3)
1-letter code: Sequence is input in format 80A1
6.2.2.3 Dihedral angle restraint information
This is the information about dihedral-angle restraints, if any are present. It is specified only when WITH_DIHED_CONSTR is present in the first record.
1st line: ndih_constr - number of restraints (free format).
2nd line: ftors - force constant (free format).
Each of the following ndih_constr lines:
idih_constr(i),phi0(i),drange(i) (free format)
-
-
idih_constr(i) - the number of the dihedral angle gamma corresponding to the ith restraint.
-
-
phi0(i) - center of dihedral-angle restraint.
-
-
drange(i) - range of flat well (no restraints for phi0(i) +/- drange(i)).
6.2.2.4 Disulfide-bridge data
1st line: NS, (ISS(I),I=1,NS) (free format)
-
-
NS - number of cystine residues forming disulfide bridges.
-
-
ISS(I) - the number of the Ith disulfide-bonding cystine in the sequence.
nd line: NSS, (IHPB(I),JHPB(I),I=1,NSS) (free format)
-
-
NSS - number of disulfide bridges
-
-
IHPB(I),JHPB(I) - the first and the second residue of ith disulfide link
Because the input is in free format, each line can be split.
6.2.3 Energy-term weights and parameter files
There are NPARMSET records specified below. All items described in this section are input in keyword-based mode.
1st record: Weights for the following energy terms:
-
-
WSC (1.0) - side-chain-side-chain interaction energy.
-
-
WSCP (1.0) - side chain-peptide group interaction energy.
-
-
WELEC (1.0) - peptide-group-peptide group interaction energy.
-
-
WEL_LOC (1.0) - third-order backbone-local correlation energy.
-
-
WCORR (1.0) - fourth-order backbone-local correlation energy.
-
-
WCORR5 (1.0) - fifth-order backbone-local correlation energy.
-
-
WCORR6 (1.0) - sixth-order backbone-local correlation energy.
-
-
WTURN3 (1.0) - third-order backbone-local correlation energy of pairs of peptide groups separated by a single peptide group.
-
-
WTURN4 (1.0) - fourth-order backbone-local correlation energy of pairs of peptide groups separated by two peptide groups.
-
-
WTURN6 (1.0) - sixth-order backbone-local correlation energy for pairs of peptide groups separated by four peptide groups.
-
-
WBOND (1.0) - virtual-bond-stretching energy.
-
-
WANG (1.0) - virtual-bond-angle-bending energy.
-
-
WTOR (1.0) - virtual-bond-torsional energy.
-
-
WTORD (1.0) - virtual-bond-double-torsional energy.
-
-
WSCCOR (1.0) - sequence-specific virtual-bond-torsional energy.
-
-
WDIHC (0.0) - dihedral-angle-restraint energy.
-
-
WHPB (1.0) - distance-restraint energy.
2nd record: Parameter files. If filename is not specified that corresponds to particular parameters, the respective name from the C-shell script will be assigned. If no files are to be specified, an empty line must be inserted.
-
-
BONDPAR - bond-stretching parameters.
-
-
THETPAR - backbone virtual-bond-angle-bending parameters.
-
-
ROTPAR - side-chain-rotamer parameters.
-
-
TORPAR - backbone-torsional parameters.
-
-
TORDPAR - backbone-double-torsional parameters.
-
-
FOURIER - backbone-local - backbone-electrostatic correlation parameters.
-
-
SCCORAR - sequence-specific backbone-torsional parameters (not used at present).
-
-
SIDEPAR - side-chain-side-chain-interaction parameters.
-
-
ELEPAR - backbone-electrostatic-interaction parameters.
-
-
SCPPAR - backbone-side-chain-interaction parameters.
6.2.4 (M)REMD/Hamiltonian (M)REMD setting specification
If HAMIL_REP is present in general data, read the following group of records only once; otherwise, read for each parameter set (NPARSET times total).
-
-
NT (1) - number of temperatures.
-
-
REPLICA - if present, replicas in temperatures were specified with this parameter set.
-
-
UMBRELLA - if present, umbrella-sampling was run with this parameter set.
-
-
READ_ISET - if present, umbrella-sampling-window number is read from the compressed Cartesian coordinate (cx) file even if the data are not from umbrella-sampling run(s). ISET is present in the cx files from the present version of UNRES.
Following NT records are for consecutive temperature replicas; each record is organized as keyword-based input:
-
TEMP (298.0) - initial temperature of this replica (replicas in MREMD).
-
FI (0.0) - initial values of the dimensionless free energies for all q-restraint windows for this replica (NR values).
-
-
KH (100.0) - force constants of q restraints (NR values). Q0 (0.0d0) - q-restraint centers (NR values)</p>
6.2.5 Information of files from which to read conformations
If HAMIL_REP is present in general data, read the following two records only once; otherwise, read for each parameter set (NPARSET times total).
1st record (keyword-based input):.
For temperature replica only ONE record is read; for non-(M)REMD runs, NT records must be supplied. The records are in keyword-based format.
-
-
NFILE_ASC - number of files in ASCII format (UNRES Cartesian coordinate (x) files) for current parameter set.
-
-
NFILE_CX - number of compressed coordinate files (cx files) for current parameter set.
-
-
NFILE_BINi - number of binary coordinate files (now obsolete because it requires initial conversion of ASCII format trajectories into binary format).
It is strongly recommended to use cx files from (M)REMD runs with TRAJ1FILE option. Multitude of trajectory files which are opened and closed by different processors might impair file system accessibility. Should you wish to process trajectories each one of which is stored in a separate file, better collate the required slices of them first to an x file by using the xdrf2x program piped to the UNIX cat command.
coordinate file name(s) without extension.
6.2.6 Information of reference structure and comparing scheme
The following records pertain to setting up the classification of conformation aimed ultimately at obtaining a class numbers. Fragments and pairs of fragments are specified and compared against those of reference structure in terms of secondary structure, number of contacts, rmsd, virtual-bond-valence and dihedral angles, etc. Then the class number is constructed as described in ref 3. A brief description of comparison procedure is as follows:
-
Elementary fragments usually corresponding to elements of secondary or supersecondary structure are selected. Based on division into fragments, levels of structural hierarchy are defined.
-
At level 1, each fragment is checked for agreement with the corresponding fragment in the native structure. Comparison is carried out at two levels: the secondary structure agreement and the contact-pattern agreement level.
At the secondary structure level the secondary structure (helix, strand or undefined) in the fragment is compared with that in the native fragment in a residue-wise manner. Score 0 is assigned if the structure is different in more than 1/3 of the fragment, 1 is assigned otherwise.
The contact-pattern agreement level compares the contacts between the peptide groups of the backbone of the fragment and the native fragment and also compares their virtual-bond dihedral angles gamma. It is allowed to shift the sequence by up to 3 residues to obtain contact pattern match. A score of 0 is assigned if more than 1/3 of native contacts do not occur or there is more than 60 deg (usually, but this cutoff can be changed) maximum difference in gamma. Otherwise score 1 is assigned.
The total score of a fragment is an octal number consisting of bits hereafter referred to S (secondary structure) C (contact match) and H (sHift) (they are in the order HCS). Their values are as follows:
-
S - 1 native secondary structure; 0 otherwise,
-
-
C - 1 native contact pattern; 0 otherwise,
-
-
H - 1 contact match obtained without sequence shift 0 otherwise.
For example, octal 7 (111) corresponds to native secondary structure, native contact pattern, and no need to shift the sequence for contact match; octal 1 (001) corresponds to native secondary structure only (i.e., nonnative contact pattern).
-
At level 2, contacts between (i) the peptide groups or (ii) the side chains within pairs of fragments are compared. Case (i) holds when we seek contacts between the strands of a larger beta-sheet formed by two fragments, case (ii) when we seek the interhelix or helix-beta sheet contacts. Additionally, the pairs of fragments are compared with their native counterparts by rmsd.
Score 0 is assigned to a pair of fragments, if it has less than 2/3 native contacts and too large rmsd (a cut-off of 0.1 A/residue is set), score 1 if it has enough native contacts and sufficiently low rmsd, but the sequence has to be shifted to obtain a match, and score 2, if sufficient match is obtained without shift.
-
At level 3 and higher, triads, quadruplets,..., etc. of fragments are compared in terms of rmsd from their native counterparts (the last level corresponds to comparing whole molecules). The score (0, 1, or 2) is assigned to each composite fragment as in the case of level 2.
-
The TOTAL class number of a structure is a binary number composed of parts of scores of fragments, fragment pairs, etc. It is illustrated on the following example; it is assumed that the molecule has three fragment as in the case of 1igd.
level 1 level 2 level 3
123 123 123||1-2 1-3 2-3 1-2 1-3 2-3 || 1-2-3 | 1-2-3 ||
sss|ccc|hhh|| c c c | h h h || r | h ||
Bits s, c, and h of level 1 are explained in point 2; bits c and h of level 2 pertain to contact-pattern match and shift; bits r and h of level 3 pertain to rmsd match and shift for level 3.
The input is specified as follows:
1st record (keyword-based input):
-
VERBOSE - if present, detailed output in classification (use if you want to fill up the disk).
-
-
PDBREF - if present, the reference structure is read from the pdb.
-
-
BINARY - if present, the class will be output in octal/quaternary/binary format for levels 1, 2, and 3, respectively.
-
-
DONT_MERGE_HELICES - if present, the pieces of helices that contain only small breaks of hydrogen-bonding contacts (e.g., a kink) are not merged in a larger helix.
-
-
NLEVEL=n - number of classification levels.
-
-
n >0 - the fragments for n levels will be defined manually.
-
-
n <0 - the number of levels is -n and the fragments will be detected automatically.
-
-
START=n - the number of conformation at which to start.
-
-
END=n - the number of conformation at which to end.
-
-
FREQ=n (1) - sampling frequency of conformations; e.g. FREQ=2 means that every second conformation will be considered.
-
-
CUTOFF_UP=x - upper boundary of rmsd cutoff (the value is per 50 residues).
-
-
CUTOFF_LOW=x - lower boundary of rmsd cutoff (per 50 residues).
-
-
RMSUP_LIM=x - lower absolute boundary of rmsd cutoff (regardless of fragment length).
-
-
RMSUPUP_LIM=x - upper absolute boundary of rmsd cutoff (regardless of fragment length).
-
-
FRAC_SEC=x (0.66666) the fraction of native secondary structure to consider a fragment native in secondary structure.
2nd record:
For nlevel <0 (automatic fragment assignment):
-
SPLIT_BET=n (0) : if 1, the hairpins are split into strands and strands are considered elementary fragment.
-
-
ANGCUT_HEL=x (50): cutoff on gamma angle differences from the native for a helical fragment.
MAXANG_HEL=x (60) : as above but maximum cutoff
-
-
ANGCUT_BET=x (90), MAXANG_BET=x (360), ANGCUT_STRAND=x (90), MAXANG_STRAND=x (360) - same but for a hairpin or sheet fragment.
-
-
FRAC_MIN=x (0.6666) - minimum fraction of native secondary structure.
-
-
NC_FRAC_HEL=x (0.5) - fraction of native contacts for a helical fragment.
-
-
NC_REQ_HEL=x (0) - minimum required number of contacts.
-
-
NC_FRAC_BET=x (0.5), NC_REQ_BET=x (0) - same for beta sheet fragments.
-
-
NC_FRAC_PAIR=x (0.3), NC_REQ_PAIR=x (0) : same for pairs of segments.
-
-
NSHIFT_HEL=n (3), NSHIFT_BET=n (3), NSHIFT_STRAND=n (3), NSHIFT_PAIR=n (3) - allowed sequence shift to match native and compared structure for the respective types of secondary structure.
-
-
RMS_SINGLE=n (0), CONT_SINGLE=n (1), LOCAL_SINGLE=n (1), RMS_PAIR=n (0).
-
-
CONT_PAIR=n (1) - types of criteria in considering the geometry of a fragment or pair native; 1 means that the criterion is turned on.
For nlevel >0 (manual assignment):
Level 1:
1st line:
-
NFRAG=n - number of elementary fragments.
Next lines (one group of lines per each fragment):
1st line:
-
NPIECE=n - number of segments constituting the fragment.
-
ANGCUT, MAXANG, FRAC_MIN, NC_FRAC, NC_REQ - criterial numbers of native-likeness as for automatic classification.
-
-
LOCAL, ELCONT, SCCONT, RMS : types of criteria implemented, as for automatic classification except that ELECONT and SCCONT mean that electrostatic or side-chain contacts are considered, respectively.
NPIECE following lines:
IFRAG1=n, IFRAG2=n - the start and end residue of a continuous segment constituting a fragment.
Level 2 and higher:
1st line:
-
NFRAG=n - number of fragments considered at this level.
For each fragment the following line is read:
-
-
NPIECE=n - number of elementary fragments (as defined at level 1) constituting this composite fragment.
-
-
IPIECE=i1 i2 ... in - the numbers of these fragments.
-
-
NC_FRAC, NC_REQ : contact criteria (valid only for level 2).
-
-
ELCONT, SCCONT, RMS : as for level 1; note, that for level 3 and higher the only criterion of nativelikeness is rms.
3rd (for nlevel <0) or following (for n >0) line:
Name of the file with reference structure (e.g., the pdb file with the experimental structure)
6.3 The structure of the main output file (out)
The initial portion of the main output file, named INPUT.out_POT_000 contains information of parameter files specified in the C-shell script, compilation info, and the UNRES numeric code of the amino-acid sequence. Subsequently, actual energy-term weights and parameter files are printed. If lprint was set at .true. in parmread.F, all energy-function parameters are printed. If REFSTR was specified in the control-data list, the program then outputs the read reference-structure coordinates and partition of structure into fragments. Subsequently, the information about the number of structures read in and those that were rejected is printed followed by succinct information form the iteration process. Finally, the histograms (also output separately to specific histogram files; see section 6.6) and the data of the dependence of free energy, energy, heat capacity, and conformational averages on temperature are printed (these are also output separately to file described in section 6.6).
The output files corresponding to non-master processors (INPUT.out_POT_xxx where xxx >0 contain only the information up to the iteration protocol. These files can be deleted right after the run.
6.4 The thermodynamic quantity and ensemble average (thermal) files
The files INPUT.thermal or INPUT_slice_yy.thermal contain thermodynamic, ensemble-averaged conformation-dependent quantities and their temperature derivatives. The structure of a record is as follows:
|
T |
F |
E |
q1…qn |
rmsd |
Rgy |
Cv |
|
298.0 |
-83.91454 |
-305.28112 |
0.30647 |
6.28347 |
11.61204 |
0.70886E+01 |
|
var(q1)… |
var(rmsd) |
var(Rgy) |
cov(q1,E) … |
cov(rmsd,E) |
cov(Rgy,E) |
|
var(qn) |
|
|
cov(qn,E) |
|
|
|
0.35393E-02 |
0.51539E+01 |
0.57012E+00 |
0.43802E+00 |
0.62384E+01 |
0.33912E+01 |
where:
-
T - absolute temperature (in K),
-
F - free energy at T,
-
E - average energy at T,
-
q1…qn: ensemble-averaged q values at T (usually only the total q corresponding to whole molecule is requested, as in the example above, but the user can specify more than one fragment or pair of fragments for which the q's are calculated, If there is no reference structure, this entry contains a 0,
-
rmsd - ensemble-averaged root mean square deviation at T,
-
Rgy - ensemble-averaged radius of gyration computed from Calpha coordinates at T,
-
Cv - heat capacity at T,
-
var(q1)…var(qn) - variances of q's at T,
-
var(rmsd) - variance of rmsd at T,
-
var(Rgy) - variance of radius of gyration at T,
-
cov(q1,E)…cov(qn,E) - covariances of q's and energy at T,
-
cov(rmsd,E) - covariance of rmsd and energy at T,
-
cov(Rgy,E) - covariance of radius of gyration and energy at T.
-
According to Camacho and Thirumalali (Europhys. Lett., 35, 627, 1996), the maximum of the variance of the radius of gyration corresponds to the collapse point of a polypeptide chain and the maximum variance of q or rmsd corresponds to the midpoint of the transition to the native structure. More precisely, these points are inflection points in the plots of the respective quantities which, with temperature-independent force field, are proportional to their covariances with energy.
6.5 The conformation summary with classification (stat) files
The stat files (with names INPUT_POT_xxx.stat or INPUT_POT_sliceyyxxx.stat; where yy is the number of a slice and xxx is the rank of a processor) contain the output of the classification of subsequent conformations (equally partitioned between processors). The files can be concatenated by processor rank to get a summary file. Each line has the following structure (example values are also provided):
|
|
|
whole molecule |
|
No |
energy |
rmsd |
q |
ang |
|
9999 |
-122.42 |
4.285 |
0.3751 |
47.8 |
|
level 1 |
|
frag 1 |
frag 2 |
frag 3 |
class 1 |
|
n1 |
n2 |
n3 |
rmsd |
q |
ang |
rmsd |
q |
ang |
rmsd |
q |
ang |
|
|
4 |
10 |
21 |
0.6 |
0.33 |
16.7 |
3.6 |
0.42 |
56.3 |
0.7 |
0.12 |
16.5 |
737 |
|
level 2 |
level 3 |
|
|
nc1 |
nc2 |
rmsd |
q |
rmsd |
q |
class 2 |
rmsd |
q |
class 3 |
class |
|
9 |
0 |
1.6 |
0.20 |
4.3 |
0.20 |
20 |
0 |
4.0 |
2 |
737.20.2 |
where
-
No - the number of the conformation.
-
"whole molecule" denotes the characteristics of the whole molecule q = 1-Wolynes'q.
-
level 1, 2, and 3 denote the characteristics computed for the respective fragments as these levels.
-
n1, n2, n3 - number of native contacts for a given segment.
-
cl1, cl2, cl3 - group of segment classes for segments at level 1, 2, and 3, respectively.
-
class - total class of the conformation.
The octal/quaternary/binary numbers denoting the class for a fragment at level 1, 2, and 3, respectively, are described in ref. [3].
6.6 The histogram files
The histogram file with names INPUT_[par_yy][_slice_xx].hist where xx denotes the number of the slice and yy denotes the number of the parameter if SEPARATE_PARSET was specified in input contain histograms of q at replica temperatures and energy-parameter sets; with SEPARATE_PARSET histograms corresponding to subsequent parameter sets are saved in files with par_yy infixes. The histograms are multidimensional if q is a vector (usually, however, q corresponds to the entire molecule and, consequently, the histograms are one-dimensional). The histogram files are printed if histfile and histout was specified in the control data record.
Each line of a histogram file corresponds to a given (multidimensional) bin in q contains the following:
The state density is printed to file(s) INPUT[_slice_xx].ent. Each line contains the left boundary of the energy bin and ln(state density) followed by "ent" string. At present, the state density is calculated correctly only if one energy-parameter set is used.</p>
6.7 The rmsd-radius of gyration potential of mean force files
These files with names INPUT[_par_yy][_slice_xx].rmsrgy contain the two-dimensional potentials of mean force in rmsd and radius of gyration at all replica-exchange temperatures and for all energy-parameter sets. A line contains the left boundaries of the radius of gyration - rmsd bin (radius of gyration first) (format 2f8.2) and the PMF values at all replica-exchange temperatures (e14.5), followed by the number of the parameter set. With SEPARATE_PARSET, the PMFs corresponding to different parameter sets are printed to separate files.
6.8 The PDB files
The PDB files with names INPUT_[slice_xx_]Tyyy.pdb, where Tyyy specifies a given replica temperature contain the conformations whose probabilities at replica temperature T sum to 0.99, after sorting the conformations by probabilities in descending order. The PDB files follow the standard format; see ftp://ftp.wwpdb.org/pub/pdb/doc/format_descriptions/Format_v33_Letter.pdf For single-chain proteins, an example is as follows:
REMARK CONF 9059 TEMPERATURE 330.0 RMS 8.86
REMARK DIMENSIONLESS FREE ENERGY -1.12726E+02
REMARK ENERGY -2.22574E+01 ENTROPY -7.87818E+01
ATOM 1 CA VAL 1 8.480 5.714 -34.044
ATOM 2 CB VAL 1 9.803 5.201 -33.968
ATOM 3 CA ASP 2 8.284 2.028 -34.925
ATOM 4 CB ASP 2 7.460 0.983 -33.832
.
.
.
ATOM 115 CA LYS 58 28.446 -3.448 -12.936
ATOM 116 CB LYS 58 26.613 -4.175 -14.514
TER
CONECT 1 3 2
.
.
.
CONECT 113 115 114
CONECT 115 116
where
-
CONF is the number of the conformation from the processed slice of MREMD trajectories.
-
TEMPERATURE is the replica temperature.
-
RMS is the Calpha rmsd from the reference (experimental) structure.
-
DIMENSIONLESS FREE ENERGY is -log(probability) (equation 14 of ref 2) for the conformation at this replica temperature calculated by WHAM.
-
ENERGY is the UNRES energy of the conformation at the replica temperature (note that UNRES energy is in general temperature dependent).
-
ENTROPY is the omega of equation 15 of ref 2 of the conformation.
In the ATOM entries, CA denotes a Calpha atom and CB denotes UNRES side-chain atom. The CONECT entries specify the C C
C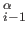 , CC
, CC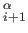 and CSCi links.
and CSCi links.
The PDB files generated for oligomeric proteins are similar except that chains are separated with TER and molecules with ENDMDL records and chain identifiers are included. An example is as follows:
REMARK CONF 765 TEMPERATURE 301.0 RMS 11.89
REMARK DIMENSIONLESS FREE ENERGY -4.48514E+02
REMARK ENERGY -3.58633E+02 ENTROPY 1.51120E+02
ATOM 1 CA GLY A 1 -0.736 11.305 24.600
ATOM 2 CA TYR A 2 -3.184 9.928 21.998
ATOM 3 CB TYR A 2 -1.474 10.815 20.433
.
.
.
ATOM 40 CB MET A 21 -4.033 -2.913 27.189
ATOM 41 CA GLY A 22 -5.795 -10.240 27.249
TER
ATOM 42 CA GLY B 1 6.750 -6.905 19.263
ATOM 43 CA TYR B 2 5.667 -4.681 16.362
.
.
.
ATOM 163 CB MET D 21 4.439 12.326 -4.950
ATOM 164 CA GLY D 22 10.096 14.370 -9.301
TER
CONECT 1 2
CONECT 2 4 3
.
.
.
CONECT 39 41 40
CONECT 42 43
.
.
.
CONECT 162 164 163
ENDMDL
6.9 The compressed Cartesian coordinates (cx) files
These files contain compressed data in the Europort Data Compression XDRF library format written by Dr. F. van Hoesel, Groeningen University (http://hpcv100.rc.rug.nl/xdrfman.html). The files are written by the cxwrite subroutine. The resulting cx file contains the omega factors to compute probabilities of conformations at any temperature and any energy-function parameters if Hamiltonian replica exchange was performed in the preceding UNRES run. The files have general names INPUT[_par_yy][_slice_xx].cx where xx is slice number and yy is parameter-set.
The items written to the cx file are as follows (the precision is 5 significant digits):
-
Cartesian coordinates of Calpha and SC sites</p>
-
nss (number of disulfide bonds)
-
if nss >0:
-
ihpb (first residue of a disulfide link)
-
jhpb (second residue of a disulfide link)
-
UNRES energy at that replica temperature that the conformation was at snapshot-recording time,
-
ln(omega) of eq 15 of ref [2],
-
Cα rmsd
-
conformation class number (0 if CLASSIFY was not specified).
7 SUPPORT
Dr. Adam Liwo
Faculty of Chemistry, University of Gdansk
ul. Wita Stwosza 63, 80-308 Gdansk Poland.
phone: +48 58 523 5124
fax: +48 58 523 5012
e-mail: adam(at)sun1.chem.univ.gda.pl
Dr. Cezary Czaplewski
Faculty of Chemistry, University of Gdansk
ul. Wita Stwosza 63, 80-308 Gdansk Poland.
phone: +48 58 523 5126
fax: +48 58 523 5012
e-mail: cezary.czaplewski@ug.edu.pl
Dr. Adam Sieradzan
Faculty of Chemistry, University of Gdansk
ul. Wita Stwosza 63, 80-308 Gdansk Poland.
phone: +48 58 523 5124
fax: +48 58 523 5012
e-mail: adasko(at)sun1.chem.univ.gda.pl
Prepared by Adam Liwo, 02/19/12.
LATEXversion, 09/27/12.
Revised by Adam Liwo, 12/04/14
This document is available in the PDF format also.
